


Pfizer/BioNTech C4591001 Trial - Audit Report - v1 (2024-05-31)
Reanalysis of the data and anomalies inventoried




Abstract
This review aims to address significant anomalies and discrepancies that have surfaced from the examination of the data from the Pfizer/BioNTech C4591001 trial, which could have profound implications for public trust and regulatory standards, should they not be adequately and transparently investigated.
Whistleblower Testimonies
Brook Jackson, a former regional director at Ventavia Research Group, reported systemic issues including lack of proper informed consent, enrollment of ineligible participants, data fabrication, and regulatory non-compliance. Despite notifying the FDA, these concerns were not adequately addressed, and critical sites were not inspected, raising questions about regulatory oversight.
Case of Augusto Roux
Dr. Augusto Roux, a trial participant in Argentina, experienced severe adverse effects after receiving the vaccine, including pericarditis, which was initially misclassified as a potential COVID-19 case at the request of the sponsor. His reports of adverse effects were inadequately documented, and attempts were made to reclassify his symptoms. Roux's case highlights serious violations of GCPs and criminal investigations are ongoing in Argentina regarding the events he witnessed.
Discrepancies in Randomization Figures
Analysis of two investigator files, produced on November 26, 2020, and March 29, 2021, revealed unexplained discrepancies in the number of randomized subjects. For instance, site 1231/4444 in Argentina showed a decrease from 5776 to 5615 subjects, amounting to 161 subjects "lost to randomization," which cannot be accounted for by subject movement between sites or recruitment of subjects under 16 years. Overall, 1203 subjects were "lost to randomization" across 108 of 153 sites, indicating a significant anomaly.
Missing Subject Identifiers
Anomalies in subject identifiers were found, with 301 missing IDs disproportionately affecting certain sites, notably in Argentina. Examination of this problem suggests non-random data handling issues, which could indicate deliberate subjects data deletion.
Delayed Reporting of Deaths
Michels et al. and further correspondence by Jeyanthi Kunadhasan with the Australian Therapeutic Goods Administration (TGA) stresses the importance of delayed reporting of deaths in the Emergency Use Authorization (EUA) figures. The Polack et al. study in the New England Journal of Medicine reported six deaths, while Pfizer's internal records already showed eight, with two deaths in the BNT162b2 arm not disclosed. These delays and non-disclosures, particularly of deaths due to cardiac events, raise serious concerns about transparency and potential breaches of GCPs.
Anomalies Related to Protocol Deviations
Protocol deviations recorded without blind review, contrary to standard clinical trial practices, showed significant disproportions in treatment groups. Notable deviations included the improper administration of protocol-specified procedures, urine pregnancy tests not performed and the receipt of other non-study coronavirus vaccines, suggesting unblinding of the participants themselves, and improper treatment by site staff.
Unreported Adverse Events
Instances of under-reported adverse events were identified, such as a case where "chest tightness" was not logged in the adverse events log but was only noted in the case report forms (CRF) due to a subsequent serious adverse event (SAE). Another example involved misclassification of a serious case of pericarditis as COVID-19 illness, highlighting violations of Good Clinical Practices (GCPs).
Adverse Effects Re-qualification
Augusto Roux’s testimony reveals systemic exploitation of protocol weaknesses, leading to widespread re-qualification of adverse effects as COVID-19 symptoms. Analysis of AESPID data indicates that at least 1209 adverse effects across 767 subjects were re-qualified to impact efficacy, safety, and immunogenicity results. This re-qualification process appears to be a deliberate manipulation to suppress adverse events in the safety analysis, as evidenced by the high proportion of communicated CRFs to the FDA among the affected subjects.
Further Evidence of Data Manipulation
Additional anomalies were identified in the recording of adverse effects. Manual reviews of CRFs revealed discrepancies such as unrecorded adverse events and events reclassified or deleted. This includes significant under-reporting and potential alteration of CRFs, violating GCPs and potentially infringing on US and international regulations.
Process 2 mis-representation and concerns
In the development of its COVID-19 vaccine, Pfizer/BioNTech utilized two distinct manufacturing methods: "Process 1" for most of the clinical trials and "Process 2" for commercial production, which was tested on only 252 recipients. Process 1 involved PCR amplification for DNA template production, while Process 2 used linearized plasmid DNA cultured in E. coli, among other modifications to scale up production. Regulatory documents from the FDA, EMA, PMDA, and TGA confirm these differences and their respective uses in clinical and commercial supplies.
The planned comparative study between the two processes aimed to assess safety and immunogenicity in participants aged 16-55 but was ultimately not conducted. Analysis of adverse event reports suggests significant differences between the processes, notably higher rates of adverse effects, including lymphadenopathy and menorrhagia, in recipients of Process 2. Several findings indicate that the manufacturing changes between Process 1 and Process 2 have impacted the vaccine's safety profile.
Introduction
The unprecedented speed of COVID-19 vaccine development and emergency use authorization (EUA) involved expedited review processes. A rigorous post-hoc analysis is essential to ensure all data was collected, analyzed, and reported according to the highest scientific standards. This is crucial for effective post-market surveillance, as accurate trial data is fundamental to identifying long-term effects or rare adverse events that may not have been apparent during the initial trials. Flawed foundational data could hinder post-market monitoring, potentially jeopardizing patient safety. Ethical standards in clinical research demand transparency and accuracy in data reporting. The current flaws and lack of transparency in the C4591001 trial data violate ethical principles such as informed consent, and potentially go against the duty to do no harm if the risk/benefit has been misrepresented.
The FDA conducted its review from November 20, 2020, to December 11, 2020, resulting in a surprisingly brief 57-page memorandum1. The seriousness of this review has been called into question2. Despite promises of transparency, evidence of multiple analyst reviews, such as emails exchanged between data analysts who would have critically evaluated the submission, as claimed by Peter Marks, remains unavailable. As we have been seeking answers to essential questions on data integrity for over a year, the present authors are well-positioned to know the diligence with which the latter's department addresses these concerns. Other regulatory bodies, such as France's “Agence Nationale de Sécurité des Médicaments et des produits de Santé” (ANSM) and Australia's Therapeutic Goods Administration, have thus far failed to adequately address concerns raised as well.
In light of the direct testimonies outlining severe infringements to Good Clinical Practices (GCPs) and the supporting data illustrating how extensive were the malpractices within this trial, there is an urgent need for a comprehensive and transparent audit of the participating sites and of the Clinical Research Organizations (CROs) responsible for data management. It is the authors' hope that the public will utilize this reproducible evidence to bring the matter to the attention of policymakers, and to the legal field shall the regulators stay inactive.
The facts presented in this report are substantiated by public documents, obtained through Freedom of Information Requests (FOI) and legal proceedings. Notably, a significant proportion of these documents comes from a Texas court order3, in a proceeding engaged by Public Health & Medical Professionals for Transparency (PHMPT4), which enabled the publication of data submitted by sponsors to the FDA for the Biological Licence Application (BLA).
Authors
The following report is the synthesis of the work, and the product of a collaborative effort, between the following authors:
Josh Guetzkow, PhD, Senior lecturer at The Hebrew University of Jerusalem (Twitter, Substack).
Jeyanthi Kunadhasan MD (UKM), MMed (AnaesUM), FANZCA MMED (Monash) (Twitter, Substack)
Brook Jackson, Clinical Research Director for the Clinical Research Organisation (CRO) Ventavia during the trial, Whistle-blower (Twitter, Website)
Christine Cotton, Bio-Statistician, ex-CEO of a CRO for 22 years (Twitter, Website)
Arkmedic (anonymous), Whistle-blower, PhD & MD (Substack)
Augusto Roux, PhD, Lawyer, University of Buenos Aires, trial participant, Whistle-blower (Twitter)
A Concerned Amyloidosis (anonymous), Clinical Researcher (Twitter, Substack)
OpenVAET (anonymous), Forensic & Data Analyst (Twitter, Substack)
The elements we advance can be verified independently and used freely. Shall a signed affidavit be required for legal proceedings, please contact us via the comments.
Method
The references to all the documents used are provided, as well as the R5 code required to reproduce every figures & charts advanced.
We downloaded (R6), extracted, and categorized (R7) the data currently available via PHMPT8. When sources are required, they are provided in footnotes.
Overview of the trial & demographics
The clinical trial had for two main objectives to establish:
the safety of the product, by presenting a positive risk/benefit ratio profile (otherwise said “saving more people than it may injure”)
the efficacy of the product to prevent symptomatic COVID-19 disease confirmed by a PCR test, in accordance with the FDA requirements.
These steps had to be completed in order to obtain the Emergency Use Authorization (EUA). Pfizer & BioNTech were the first of the competing pharmaceutical groups to announce - by a press release on November 9, 2020 - that these objectives had been achieved.
The EUA was applied to on November 20, 20209, after the data had been analyzed with a data collection stop date (cut-off) on November 14, 2020. The goal was then to obtain the BLA - a goal achieved on August 23, 202110 - following an application for which the data had a cut-off on March 13, 2021.
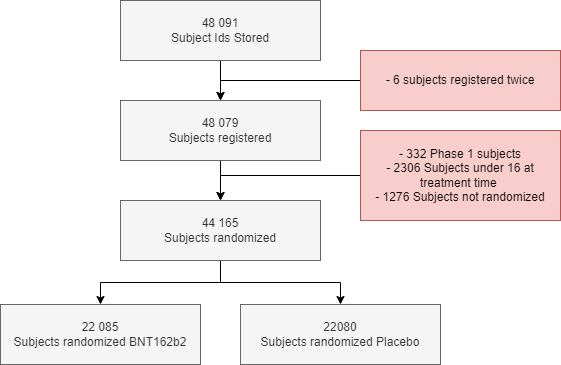
A total of 48,091 unique subject identifiers are registered in the clinical trial database, and were screened by 153 sites between April 29, 2020 and January 12, 2021 (R11).
Within the study, each of the trial sites was given a 4-digit numerical identifier, starting from 1001. One of the sites, the Dr-Mayor Cirujano Military Central Hospital in Argentina, was split into two identifiers, 1231 & 4444 (a virtual reference12). The names and addresses of the sites, as well as the identity of the principal investigator for each site, are provided in one of the documents submitted to the FDA13.
Subjects were recruited over a period of 10 weeks for phase 1 and 22 weeks for phase 2-3. The following diagram illustrates the recruitment carried out each week (R14).

The geographical coordinates of the sites were manually integrated into a JSON file15 including latitudes and longitudes. A summary of the recruitment carried out for each country was generated (R16), as well as the map below by site (R17).


A trial presented as "Placebo-controlled, blinded observer, which established safety and efficacy
The clinical trial was presented to the public as a “Phase 1/2/3, placebo-controlled, randomized, blinded, dose-ranging study to evaluate the safety, tolerability, immunogenicity and potential efficacy of SARS-CoV-2 RNA vaccine candidates against COVID-19 disease in healthy individuals”18.
Various measurements (specific neutralizing antibodies (N-Binding), PCR tests (PCR), immunoglobulin G specific to the S1 subunit of the epi and to the receptor binding domain (RBD), etc.) were carried out on subjects in phases 1 and 2-3 to determine whether they had contracted COVID, and how they reacted to the product.
Efficacy was measured on the product's ability to prevent confirmed cases of COVID-19 and was based on two primary endpoints. The primary efficacy objective was to prevent symptomatic laboratory-confirmed cases of COVID-19 at least 7 days after the second dose, in participants with no serological or virological evidence of prior SARS-CoV-2 infection, before and during the vaccination schedule. The secondary objective was to evaluate the efficacy of the vaccine against severe cases of COVID-19.
Safety objectives included assessment of local reactions, systemic events and use of antipyretics/pain medication from Day 1 to Day 7 after each dose in a subset of participants. In addition, the trial aimed to monitor unsolicited, non-serious adverse events from the first dose until one month after the second dose in all participants, serious adverse events from the first dose until six months after the second dose in all participants, and deaths and related serious adverse events from the first dose until the end of the study in all participants19.
Inconsistent dates for Phase 1
According to two studies20 21 reporting phase 1 results, the phase 1 was initiated on May 4, 2020, and the recruitment (screening) would continue until June 22, 2020.
These dates are incorrect, and the screening took place between April 29, 2020 and June 29, 2020 (R22).
Phase 1 - Products tested and demographic considerations
332 subjects were screened for phase 1. 83 failed to satisfy the screening conditions, and 54 weren’t assigned to a treatment group. Phase 1 was testing dosage and primary safety concerns, on cohorts of 15 subjects (12 being administered the treatment and 3 being administered Placebo). The treatments originally planned to be tested were:
BNT162a1 (RNA-LNP vaccine utilizing uRNA and encoding the RBD): 3 μg, 10 μg, 30 μg
BNT162b1 (BNT162 RNA-LNP vaccine utilizing modRNA and encoding the RBD): 10 μg, 30 μg, 100 μg
BNT162b2 (BNT162 RNA-LNP vaccine utilizing modRNA and encoding the P2 S): 10 μg, 30 μg, 100 μg
BNT162c2 (BNT162 RNA-LNP vaccine utilizing saRNA and encoding the RBD): 3 μg,10 μg, 30 μg
The other subjects were to receive their second dose 22 days after dose 1 - in fact a window of 19 to 23 days23 after dose 1. This window would, at EUA time and without adequate protocol amendment - being simply mentioned in the Statistical Analysis Plan (SAP), evolve to 19 to 42 days24.
Phase 1 took place at 4 sites. Sites 1001, 1002, 1003 and 1007, located in the United States, were the only ones involved (R25) - represented in the map below (R26).

The number of products tested was lower than initially planned:
24 subjects received BNT162b1 (10 mcg)
12 subjects received BNT162b1 (20 mcg)
24 subjects received BNT162b1 (30 mcg)
24 subjects received BNT162b2 (10 mcg)
24 subjects received BNT162b2 (20 mcg)
24 subjects received BNT162b2 (30 mcg)
12 subjects received a dose of BNT162b1 (100 mcg), followed by a dose of BNT162b1 (10 mcg)
39 subjects received placebo
BNT162a1 and BNT162c2 were not tested. Changes in the number of subjects scheduled for phase 1 occurred repeatedly27, while Phase 1 was ongoing:
from 840, on April 17, 2020, to 420, on June 11, 2020
from 420 to 630, on July 1st, 2020
from 630 to 195, on July 24, 2020
Pathway of subjects in Phase 1
The informed consent form28 details the subjects' journey through the phase 1 study, which involved a total of 10 planned visits.
At the first visit, the subject underwent an extensive set of tests, including measurements of their antibody levels to ensure they had not contracted COVID-19 prior to participating in the study. The subject's medical history was also reviewed, and they were confirmed to be fit to take part. After this screening process, the subject was then randomized and given either a dose of the placebo or the active product.
The following day, at the second visit, the subject's status was confirmed, and fewer tests were carried out. Notably, this was the only visit where the subject was not tested for antibodies.
The subsequent visits occurred at regular intervals. The third visit took place one week after the first dose, while the fourth visit coincided with the administration of the second dose, which was given 19 to 23 days after the first dose. The fifth and sixth visits followed at one week and two weeks after the second dose, respectively. The seventh visit occurred one month after the second dose, and the eighth visit was scheduled six months later. The ninth visit was planned for one year after the second dose, and the tenth and final visit was set to take place two years after the second dose.
BNT162b2 100 μg subjects were assigned to a dedicated 'dosing schedule', with a second dose of 10 μg administered 85-105 days after dose 1. Nowhere in the publicly available versions of the protocol is it documented why the 100 μg were subjected to such special treatment, when this should have been explicit.
The visits for which antibody measurements have been retained, at the cut-off date of March 13, 2021, for phase 1 subjects are visits 1 to 8 (R29).
Anomalies in Phase 1 antibodies measurements
Some of the antibody measurements made by subjects during Phase 1 are presented in the previously cited study by Edward Walsh et al, “Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates”30.
The ADVA file, which contains the results of these tests, shows 115 discrepancies31 between measurements taken on the same subject, during the same visit, on the same day and during the same test. In the event of a conflict, the higher value was most often maintained. Although the Analysis data reviewer guide (ADRG32) mentions this anomaly, it does not provide a reason for it. There are two mNeongreen virus neutralization assays in a document listing analytical methods33, which differ insofar that one has a limit of quantification (lower and upper bounds for minimum-maximum titer measurable). The documents referred to, VR-MQR-1021434 and VR-MVR-1008335, are the “Qualification” and “Method Validation of the SARS-CoV-2 mNeonGreen Virus Microneutralization Assay”, with submission dates of August 18, 2020 and February 9, 2021 respectively. While it seems, the difference after retesting are due to “validated” neutralization assay parameters, the experts we consulted were unable to explain how samples taken from the same subject on the same day could produce such significant differences in measurements - which raises the possibility that the tests with 'disappointing' results had been 'redone', by replacing the samples from defective subjects with samples with known high antibody levels. Admitting the method change would be valid, it would in any case raise questions on the validity of previous measurements.
As an example, the chart below illustrates the differences in measurements for the "SARS-CoV-2 serum neutralizing titer 50 (titer) - Virus Neutralization Assay" when comparing the average of the first measures made versus the average of the measures sustained.

Pathway of subjects in Phase 2-3
The informed consent form details the subjects' journey through the phase 2-3 study36, which involved a total of 6 planned visits.
At the first visit, the subject underwent a long list of tests, although fewer than those required for the phase 1 subjects. This included measurements of the subject's antibody levels to ensure they had not contracted COVID-19 prior to participating in the study. The subject's medical history was reviewed, and they were confirmed to be fit to take part. The subject was then randomized and given either a dose of the placebo or the active product sustained following phase 1, the BNT162b2 30 mcg.
The second visit was for the administration of the second dose. Notably, this was the only visit where antibody measurements were not made, which could have provided confirmation of infections that may have occurred in the days following the first dose while being asymptomatic or evading PCR detection, due, for example, to the subject not reporting symptoms, or not being tested while reporting symptoms.
The third visit took place one month after the second dose, while the fourth visit was scheduled for 6 months after the second dose. The fifth visit followed one year after the second dose, and the sixth and final visit was planned for 2 years after the second dose.
Discrepancies in reported randomization figures
Two “investigator files” were produced by the sponsors to report on the sites’ investigators and the total number of subjects screened and randomized at each site. A first document was produced on November 26, 2020 37- and a second document was produced on March 29, 202138. The first document mentions a query date of November 19, 2020, while the second mentions a query date on March 19, 2021. While the total number of subjects screened per site does not show a greater shift than the number of subjects who changed site, the total number of subjects randomized falls significantly (R39), without explanation. Subjects cannot be "de-randomized" in a clinical trial.
For example, illustrated below, site 1231/4444 in Argentina went from 5776 randomized subjects on November 26, 2020 to 5615 (-161) on March 29, 2021.
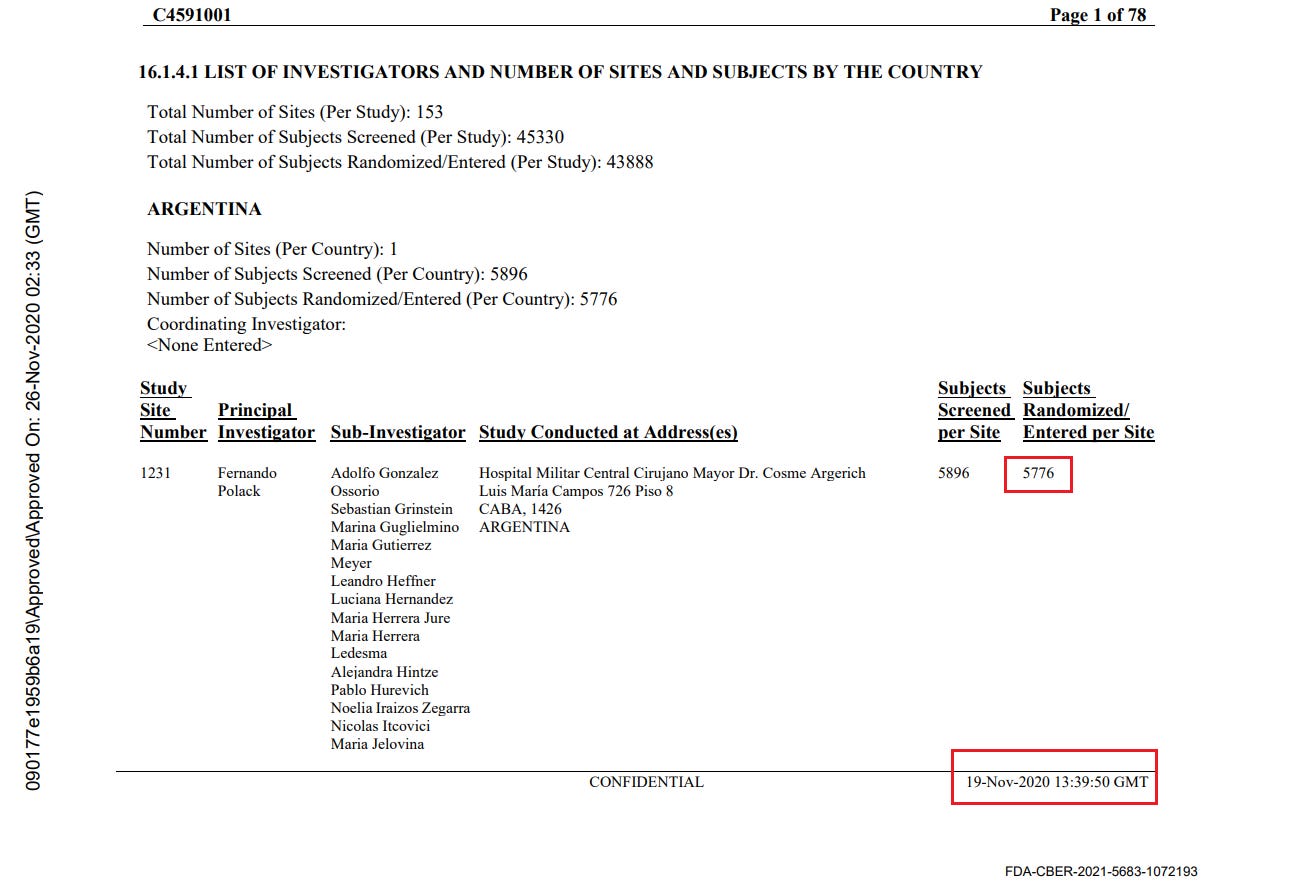
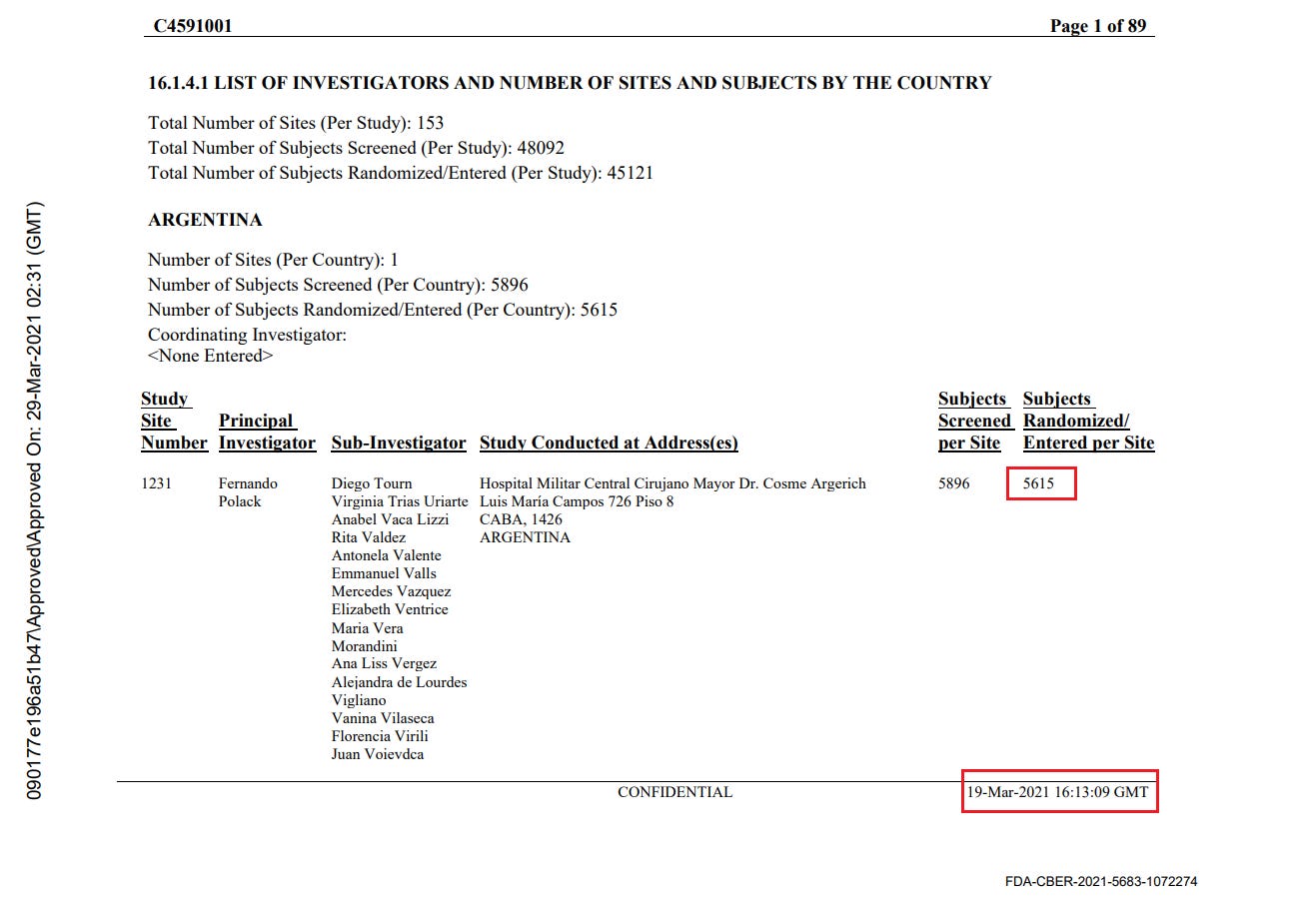
The total number of sites where subjects dropped out was 1203 subjects “lost to randomization”, over 108 of the 153 sites. This is a major anomaly which cannot be explained either by the movement of subjects between sites, or by the recruitment of subjects under 16 years of age which would have been included in one report and not the other. To illustrate this anomaly, site 1018 lost 30 subjects, while no subjects went in or out, and that no adolescents under 16 had been recruited on November 19, 2020, the date of the first file’s query - or at any time later. The excerpt below illustrates all the sites which went backward in randomization figures40.
Phase 2-3 - Randomization figures officially released
The ADRG41 documents 6 subjects who registered on more than one site, generating 12 subject identifiers, and who are subjects to specific exclusions (10561101, 11331382, 11101123, 11331405, 11491117, 12691090, 12691070, 11351357, 11341006, 10891112, 11231105, 10711213).
The BLA application memorandum42 and the corresponding Thomas et al. NEJM study, “Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months”43, both report a total of 44,165 randomized subjects as of March 13, 2021. This population has been reproduced (R44) and its conformity with the announced demographic characteristics has been verified45.
Anomalies related to the Blind in deviations
Patients whose behavior deviated from that envisaged by the protocol, or who were the subject of an error by the team in charge, had their protocol deviations recorded, which were then reviewed by a dedicated team, in order to determine whether this deviation was serious enough to justify their exclusion from a given 'population' (group dedicated to the analysis of a trial objective - e.g. efficacy).
The study protocol46, approved by the regulatory authorities, introduced a major departure from standard clinical trial practice: the on-site staff responsible for examining deviations from the protocol, to determine the eventual exclusion of participants, were not subject to blind review.
Analysis of the deviations generated by the randomized phase 3 population (R47) highlights significant disproportions in the treatment groups. The following table summarizes all the deviations observed in more than 100 subjects in the study and presenting statistically significant imbalances, when compared to the remaining of the randomized population (22 085 BNT162b2 & 22 080 Placebo), using a chi-square test.

Some very significant disproportions in the deviations - such as "Receipt of any other non-study coronavirus vaccine at any time prior to or during the study" - show that many subjects were aware that they had received a Placebo - and had enrolled in a competitor's study (probably Moderna) in parallel, or had received another product after Emergency use authorization. On the other hand, certain disproportions indicate that the teams at certain sites did not treat patients in the way expected in a blinded observational trial, where both groups should be treated equally by the clinical team:
Assessment of acute reaction for protocol specified time-frame after study intervention administration not performed at the vaccination visits
Nasal swab not collected by site staff prior to vaccination
Nasal swab not collected for the visit where it is required
Procedure/Test not performed per protocol
Urine pregnancy test not performed
It is worth noting, regarding the deviations “Receipt of any other non-study coronavirus vaccine” - that a large portion of them happened after December 2020, while other vaccines became available.
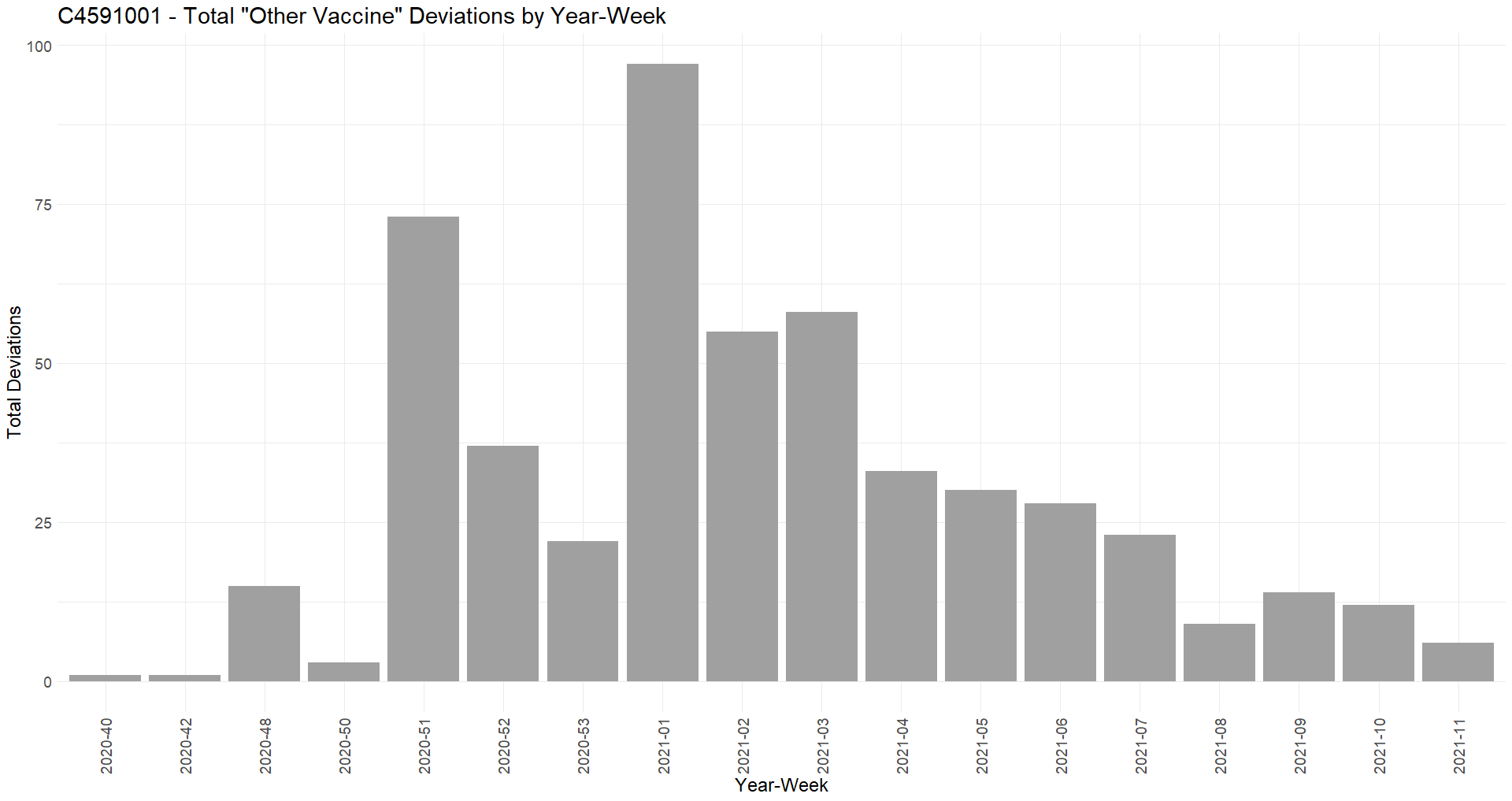
The 10 sites concerned by 20 or more of these deviations and showing significant imbalance when tested with a Fisher exact test have been isolated and synthesized in the following table.

Contrarily to the “subjects related” deviations envisaged on point 32, examining the “staff-related” deviations detailed on point 31 are in majority occurring prior to the official unblinding of the subjects.
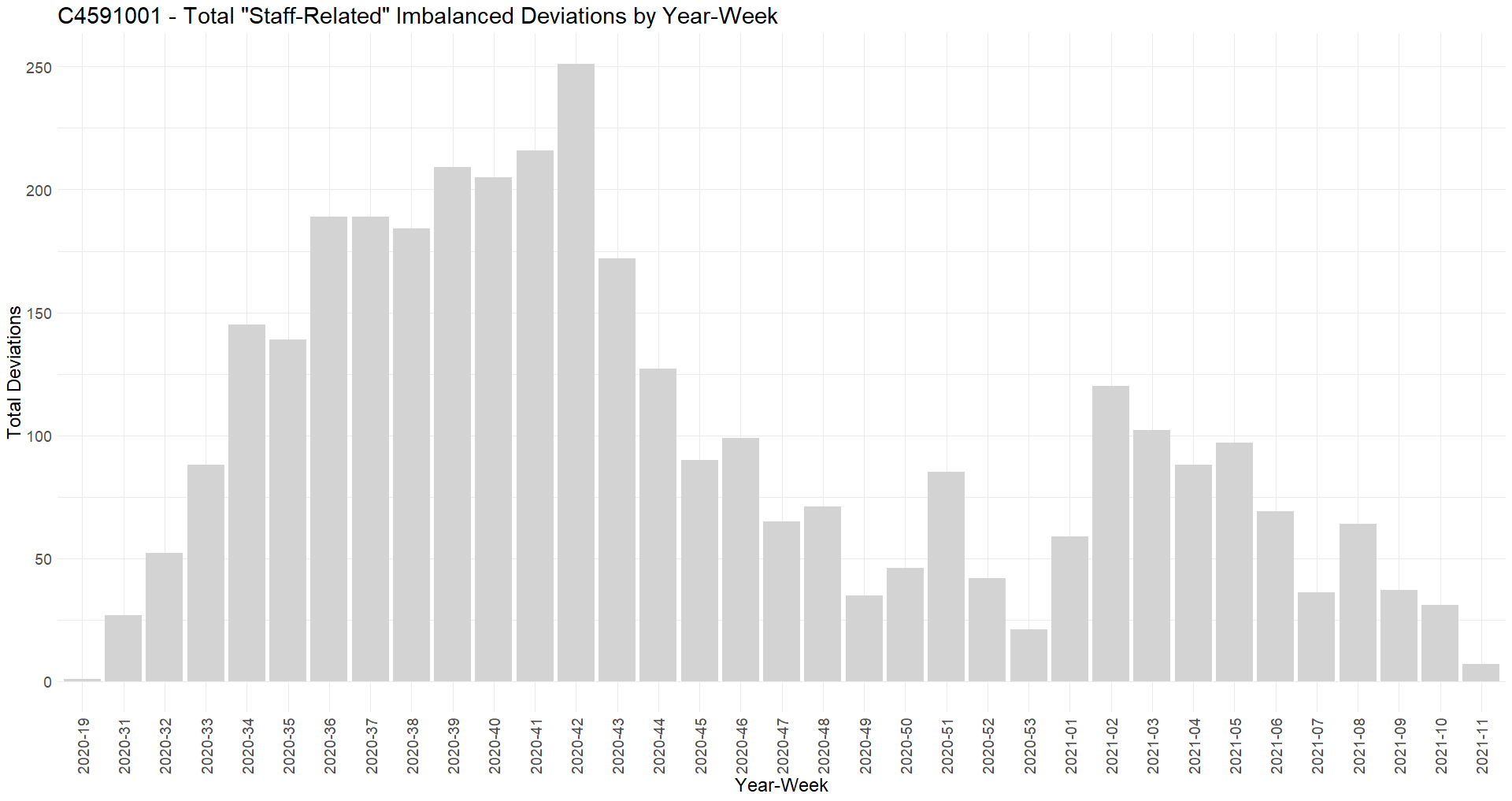
COVID visits and symptoms anomalies
When subjects experienced COVID symptoms from a list predetermined by the FDA, they had to go to the trial site to have samples taken - or, failing that, to test themselves at home with tests supplied and send the sample to the trial site, within four days48 before or after the onset of symptoms. The “official” COVID symptoms were the following 13 symptoms49:
Chills
Diarrhea
Fever
New loss of taste or smell
New or increased cough
New or increased muscle pain
New or increased shortness of breath
Sore throat
Vomiting
In addition to these symptoms, there were “non official” symptoms:
Fatigue
Headaches
Nasal congestion
Runny nose (rhinorrhea)
The “Covid-19 Signs and Symptoms” .XPT file (ADSYMPT50) however lists a total of 15 symptoms - the ones not scheduled to the protocol being marked by a (*):
Chills
Diarrhea
Fatigue
Fever
Headache
Nausea (*)
New loss of taste or smell
New or increased cough
New or increased muscle pain
New or increased nasal congestion
New or increased shortness of breath
New or increased sore throat
New or increased wheezing (*)
Rhinorrhea
Vomiting
Sites also had the ability to enter “other” symptoms, which then did not make it into the database. An example is subject 1016128951, male of age 17, BMI 22.4, who received dose 1 of BNT162b2 30 mcg on September 18, 2020, and dose 2 on October 7, 2020. He had a COVID illness entry on October 7 and 11, with assessment on October 9 and symptoms: fever, cough, muscle pain, sore throat, rhinorrhea. Page 270 of the Case report forms (CRF) however shows “chest tightness” as an “other” symptom. It is not reported in the Adverse Events (AE) log, and only visible because the young man had a serious adverse event (SAE) later, on October 31, 2020 : a motor vehicle accident where he suffered facial fractures and a concussion, which led to his CRF file being released. This is a violation of GCPs - among others the EMA Guideline for good clinical practice52.
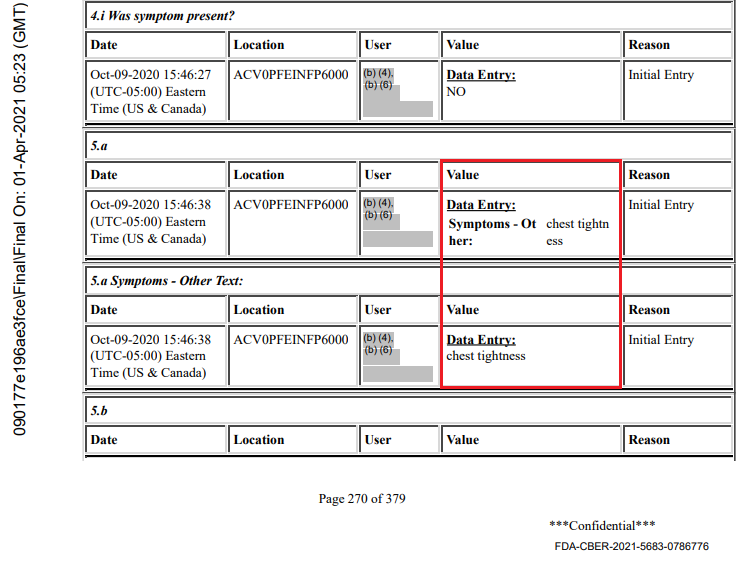
Another example of GCPs infringement by the sites is subject 1231563253, another BNT162b2 30 mcg recipient. His COVID illness entry spans from October 17, 2020 to December 7, 2020, and includes two hospitalizations. While his condition is initially described by the site as pneumonia, the subject suffered a SAE (pericarditis) which was only reported at the sponsor’s request.
The site initially reported “pneumonia” as adverse event, and later deleted it and put it into a COVID illness visit. Had Pfizer not insisted on an AE entry of the pericarditis in the CRF, it would have remained unreported in the trial data, only existing in Pfizer’s fax-based safety database.

Non-respect for the Blind and for Good Clinical Practices, testimony by Brook Jackson
A whistleblower, Brook Jackson, who served as the regional director at Ventavia Research Group (Ventavia), a site management organization responsible for enrolling participants in the trial, provided the British Medical Journal (BMJ) with internal company documents, photos, audio recordings, emails, and other evidence raising significant concerns regarding data integrity and the safety of trial participants54.
Among the systemic trial conduct issues documented across the trial sites, she reported the following:
Lack of Proper Informed Consent: During the internal quality assurance review of hundreds of individual participant records, informed consent was not obtained from each participant prior to being injected with the investigational product. In addition, it was recorded that many of these forms had obvious mismatches in signatures indicating forgery of patient signatures.
Enrollment of Ineligible Clinical Trial Participants: Clinical trial participants who did not meet Pfizer’s protocol-defined inclusion criteria, or who met exclusion criteria, were enrolled in the trial. This included pregnant women, Ventavia employees, family members and friends.
Data Fabrication and Data Falsification: Documents indicated that data points were fabricated and falsified. Examples include:
Altering patient records and backdating documents.
Forging doctor signatures.
Fabrication and falsification of missing or inconsistent data, including medical history, concomitant medication use, physical exams, vital signs, blood collection and specimen processing times, urine pregnancy testing, and other essential trial data.
Regulatory Non-Compliance/Other Protocol Violations
Failure to maintain the "blind" as required.
Protocol required study visits, including potential COVID-19 illness visits not done or completed outside of the specified timeframe.
Failure to monitor participants’ use of the electronic diary application used to record local reaction, systemic events, and use of any antipyretic medication usage following injection.
Failure to elicit, collect, and report adverse events and serious adverse events, including the follow-up of the event or its sequelae resolved.
Investigational product dosing and administration errors.
Failure to maintain proper temperature control of the investigational product.
Mislabeling and mix-up of participant laboratory specimens.
Use of unqualified and untrained research staff, including blinded vaccinators and laboratory personnel.
Principal Investigator oversight failures.
Inadequate emergency response protocol in the event of an acute allergic reaction.
Failure to assess acute reactions post vaccination.
Failure to report serious adverse events, protocol, and regulatory violations to Pfizer or the external Institutional Review Board.
Failure to report and follow-up on investigational product exposure during pregnancy, breastfeeding, and occupational exposure.
Other ethical violations, including violation of HIPAA and providing unapproved compensation to avoid participant complaints to the FDA, or news media outlets.
After repeatedly notifying Ventavia of these concerns without any action being taken to protect the rights, safety, welfare, and private health information of the trial participants she filed a complaint with the Center for Biologics Evaluation and Research (CBER), the department within the FDA that regulates biological products for human use under applicable federal laws, including the Public Health Service Act and the Federal Food, Drug and Cosmetic Act. Within hours of notifying CBER, she was terminated by Ventavia.
The FDA's Bioresearch Monitoring (BIMO) is a program involving on-site inspections, data audits, and remote regulatory assessments. It is designed to oversee all aspects of conducting and reporting FDA-regulated research. The BIMO program aims to ensure the quality and integrity of data submitted to the FDA for new product approvals and marketing applications while also protecting the rights and welfare of human subjects involved in research55. Despite receiving a complaint reporting significant violations, the FDA's BIMO program failed to inspect these critical locations. Instead, inspections were conducted at six other clinical investigator sites participating in the study. According to the FDA’s review memo, those inspections did not reveal problems impacting the data submitted in support of the original EUA.
Testimony by Augusto Roux - Non-recording and re-qualification of adverse effects
One of the participants in the clinical trial in Argentina, Dr Augusto Roux, a lawyer, is also among the authors of the present report. He reported, during the trial, several major breaches of GCPs and patient safety. The events were covered, in details, by Dr. David Healy, a University Professor in Canada & Physician, via his website56 - then in a peer-reviewed paper, “The coverage of medical injuries in company trial informed consent forms”57.
Augusto Roux volunteered for the trial, where he was given the subject number 12312982, and received his first dose on August 21, 2020. He received his second dose on September 9, 2020, at 6 pm. After his second dose, he felt very unwell an hour and a half later, collapsed once home, and was two days later diagnosed at the Alemán Hospital with pericarditis, which was judged causal to the vaccine he had received by several medical experts : aside for Dr. David Healy, Dr Gemma Torrell, a family medicine specialist from Barcelona, and Professor Joan-Ramon Laporte, one of the leading Spanish experts in pharmacovigilance.
Augusto Roux contacted the FDA, EMA & other regulators - which refrained from considering his case with the seriousness it required. He initiated a criminal investigation in Argentina, against Fernando Polack and others, for falsification of public documents, abandonment of person.
The expertise provided by his physician is attached to the present report58. These three experts have, among other major infringements to GCPs, reported that :
His side effects were not reported correctly - and some were subsequently reclassified as a potential COVID, at the request of the sponsor, BioNTech.
His pericarditis, for example, was simply not reported (R59).
Clinical trial records communicated to his physicians were containing a mixture of samples collected from several patients, weren’t chronologically ordered, and other infringements to good practices.
The records communicated to the physicians seemed scrambled, and incomplete.
There are evidences from the records that Dr Fernando Polack, without expertise in that area, attempted to have Augusto Roux classified as suffering from mental illness, and to conceal the relation between the injury and the vaccine.
Phone recordings suggest that deaths, which would have occurred during the trial, would have been concealed.
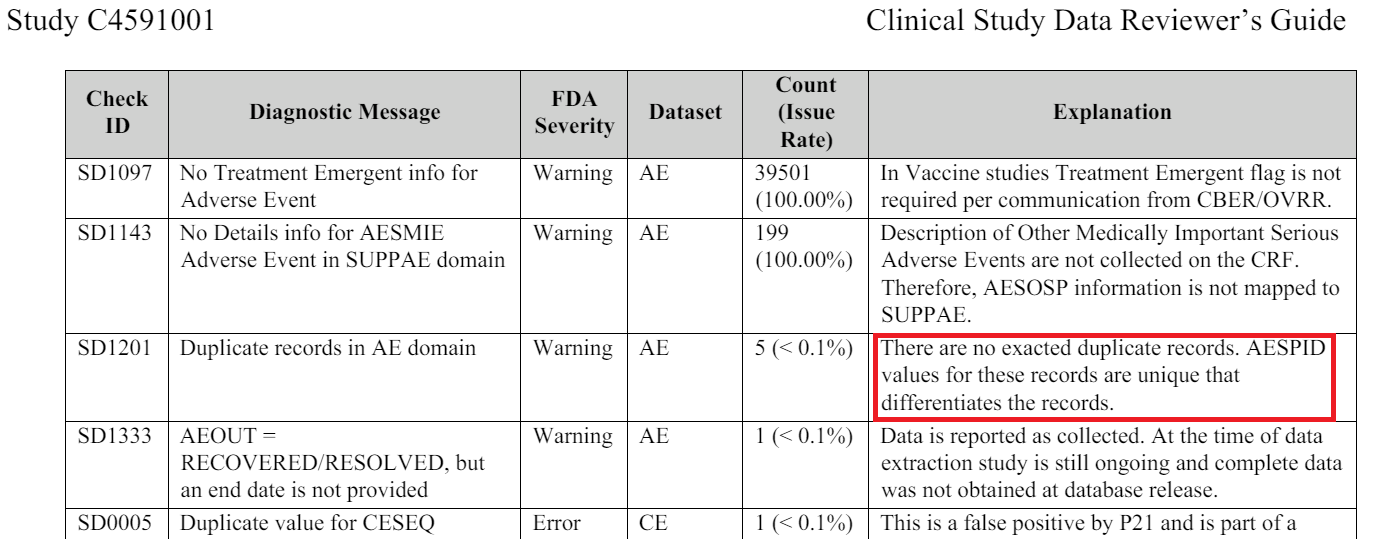
Additional indicators for deletion of subjects in base, 301 missing subjects identifiers
When a subject was registered, the ICON software (“Firecrest”, an interface synchronized by a central Oracle database) assigned it a unique and, above all, incremental identifier ("subject id" - scalar "SUBJID" in the .XPT files):

The first four digits denote the trial site identifier and are always the same for all subjects at the same site. (In some cases, subjects changed trial sites during the course of the study, so the full unique subject identifier provided information on both the 'current' and 'original' site).
The last four digits designated the order of registration, starting with 1001. The next subject registered at a given test site was then numbered 1002, then 1003, and so on.
The study identifier is circled in blue (always C4591001), the current site identifier is circled in green (here, 1016). The initial registration site is circled in yellow. The incremental subject identifier for this given site is circled in red.
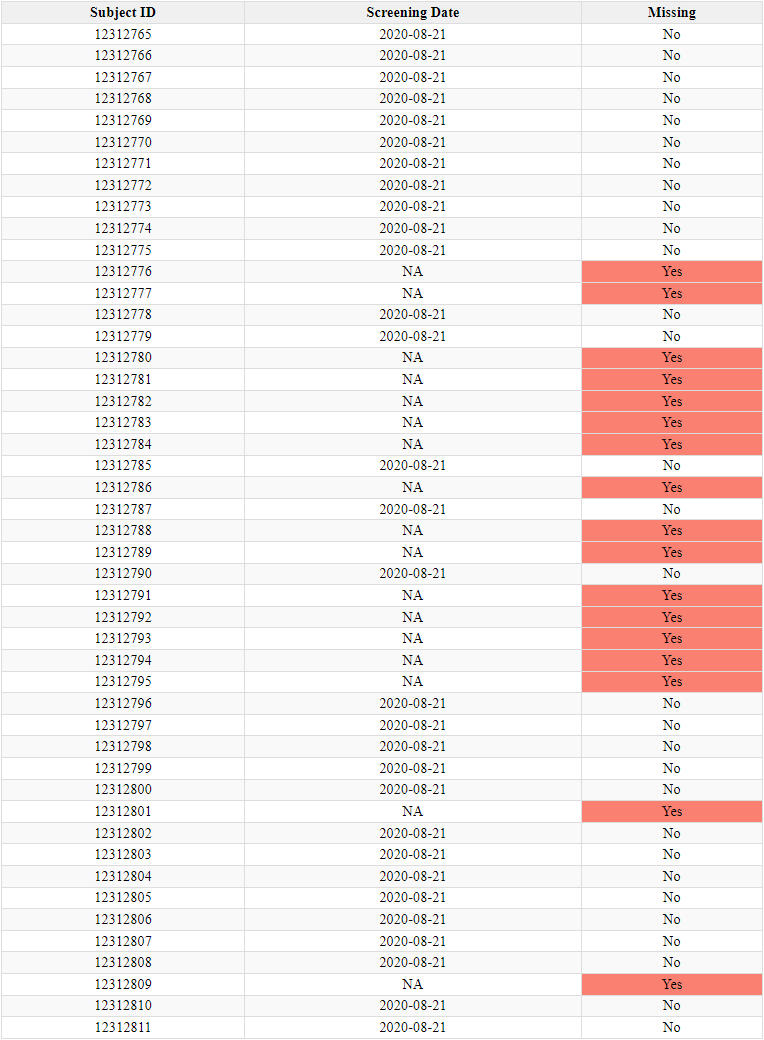
An analysis (R60) of these numbers - which are normally perfectly sequential - shows that 301 subjects are missing from the reports submitted. This anomaly disproportionately affects Argentina, where the only site represents 111 of these 301 missing subjects.


17 of these subjects - one of the most significant difference on a single site and a single day - were recruited on the same day as Augusto Roux, August 21, 2020.
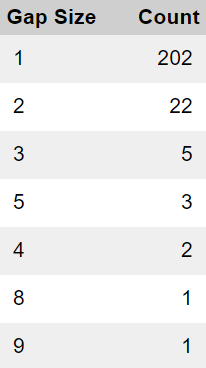
If the missing subject IDs were due to random error, we would anticipate a uniform distribution of missing IDs across all sites. However, our analysis reveals a non-random pattern, with a disproportionate concentration of missing IDs at a limited number of sites. Furthermore, we would expect to observe isolated missing ID numbers, but instead, we frequently encounter clusters of consecutive missing IDs, with gaps ranging from 2 to 9 or more consecutive ID numbers. The following breakdown summarizes the frequency of gaps of varying sizes.
Further, if these missing subject identifiers were due to some computer error (no matter how improbable it is given that we’re very far from volumes which would affect an Oracle database cache), we would expect them to be occurring on the most loaded days. We can represent the daily screening registered on the site 1231, and the subjects who may have disappeared on the recruitment dates we deduced, to illustrate the fact that August 21, 2020 wasn’t the most loaded day of the subjects enrollment on this site.

Inconsistencies in interim efficacy analyses - announcement dates & non compatible subject
The SAP61 - scheduled an efficacy analysis at 164 cases for application to the EUA, and intermediate efficacy analyses at 62 and 92 cases - at which stage efficacy could be announced if achieved.
The 170 “official” efficacy cases, presented as those obtained at the stop date of 14 November 2020, are listed in one of the .PDF files provided62. The cases don’t include their central PCR date (when Pfizer had knowledge & confirmation of the case). Extracting these cases (R63) allows for their reanalysis (R64).
It also shows that Pfizer already had the necessary 63 cases on October 22, 2020, and 94 cases on October 30. Nevertheless, they waited until November 9, 2020 to announce the results of the interim efficacy analysis via a press release. It's unclear why they delayed announcing the product's claimed efficacy so long. Perhaps does it has a relation with the fact that getting a vaccine was a central issue in the US election on November 3, 2020 - and the desire expressed by Albert Bourla65, Pfizer’s CEO, not to further politicize the discussion.

Deviations among the EUA efficacy cases
As highlighted by Dr Jeyanthi Kunadhasan, in an article for the Australian’s The Spectator66, reporting on her work with the DailyClout’s team 3, several important protocol deviations have impacted the subjects included in the EUA official cases:
Of the 170 participants, five (4 BNT & 1 Placebo) failed to receive their second dose within the protocol-defined window of 19 to 23 days. As mentioned point 13, the 19-42 day window was only mentioned in the EUA documentation (R67).
One participant (Placebo) did not receive the correct dose of the product.
Another participant (Placebo) received a blood product within 60 days, constituting a major protocol deviation.
One participants (44441224, Placebo68) withdrawn from the trial prior to the EUA issuance date but was still included in the 170.
Antibodies measurements & anomalies to the Blind
Anti-N antibodies were examined via tests that respond to a molecule within the SARS-CoV-2 virus known as the nucleocapsid (N). Anti-N antibodies are of particular interest, because the only known method for a subject to produce them is through contracting COVID-19. The N-antibodies were measured during the trial via a mNeonGreen SARS-CoV-2 “Nucleoprotein-Binding Antibodies Assay”69. As the name suggests, this test targets the Nucleoprotein, which is a component of the nucleocapsid70. It’s worth noting that N-antibodies aren’t a perfect detection method, with 5.5% of the subjects testing negative at 100 days according to observational studies71.
Placebo subjects were significantly more likely not to attend the Visit 3 or not to have their antibodies tested upon visit 3, as shown by the antibodies testing via N-binding (R72).

21 of the 1178 missing visit 3 BNTs have a positive central PCR (1.78%), and 105 of the 1392 Placebo subjects missing have a positive PCR (7.5%) (Visit 3 was performed between 46 and 138 days post visit 1). Detection of COVID appear to have been a factor in subjects not having his scheduled visit 3. But it doesn’t suffice to explain the offset - as even if we exclude these positive PCRs, it remains a significant difference.

21 191 BNT & 21 191 Placebo subjects therefore started with negative PCR & N-Binding Results at visit 1, and received their first dose. 19 928 BNT & 19 511 Placebo were negative on visit 3 N-Binding Antibodies testing, and 85 BNT subjects were positive, against 288 Placebo. 53 BNT cases were detected via Central PCR, along with 201 Placebo cases. It leaves us with 32 undetected cases among BNT subjects (37.6% of the 85 cases), compared to 87 undetected cases in the placebo group (30.2%) - illustrating how flawed was the PCR detection and how regrettable it is not to have measured antibodies at visit 2. Subjects in the BNT groups were 7.4% more likely to avoid PCR detection, while having positive COVID specific antibodies at the 1 month post dose 2 visit.
In the Comments (CO.xpt) file, there are 9113 comments associated with COVID convalescent visit blood draws (identified by RDOMAIN=IS and VISIT=COVID_n), and 8869 covid convalescent visits are in the Subject visits (SV.xpt) file (identified by VISIT=COVID_n). However, there are only 91 results associated with COVID convalescent visits in the IS.xpt (Immunogenicity) database (identified by VISIT=COVID_n1). The protocol allows for visits to overlap; if one of the visits was a convalescent visit, the samples were to be entered there. The convalescent serology remained unreported, and while the explanation for missing Visit 3 may reside in Placebo subjects who had lost interest in the trial, this represents a protocol flaw which allowed data not to be reported.
The adverse effects to COVID visit re-qualification was widespread
Augusto Roux’s testimony, points 43 to 46, is evidence that the weakness in the protocol - consisting in confusing COVID symptoms & Adverse effects from the product, along with the capacity to qualify one or the other to affect efficacy, safety and immunogenicity results - was exploited.
Aside for a single row on two subjects (10681017 & 10681036), all the Adverse Events (AEs) entries have 1 to N entries related to a subject by a unique incremental “AESPID” entry73, documented in the CSDRG74 as the “Sponsor’s ID set for the specific AEs”. The sponsors document 5 exceptions had been raised on March 13, 2021, as duplicate ids were entered by sites in this document.
By analyzing the data structure to detect gaps in these AESPID by subjects, we can detect adverse events that were deleted and suppressed from the safety analysis: a minimum of 1209 adverse effect have been re-qualified (R)75, over 767 subjects. Adverse events at the end of the log evade detection.
For a limited number of subjects (1028 subjects total - 982 in individual files), the CRF have been communicated to the FDA along with the BLA application. 97 of these individual CRFs are those of subjects among these 767 subjects (12.65%). This very significant rate of CRFs available, while only 2% are available for the 44K+ population, might be explained by the FDA having questions about part of these subjects specifically - or simply their compliance with the conditions established to the CRFs to be provided, detailed in the meetings correspondence76.

The subject’s audit trail allows us to identify which adverse effects were suppressed after being registered as the events were “re-qualified” to COVID visits. The case by case review of all the CRFs anomalies will be the subject of a later analysis - but the followings are an illustration of the severity of the phenomenon and are stressing the need for an in depth audit of the individual CRFs. Subject 1218100177, a 32 years old female, reported Diarrhea on November 4, 2020. On March 29, 2021 (16 days after the BLA application cut-off and 3 days prior the CRFs cut-off), this adverse effect is re-qualified to a “COVID visit” retroactively created under instruction from the sponsors.

Subject 1087128678, a 71 years old female, reported on October 20, 2020 a wide range of adverse effects - judged “non serious” by the PI (page 648-649). These events were re-qualified as COVID symptoms on March 29, 2021.

Further evidence of data manipulations on safety data recordings
Another important anomaly in the recording of the adverse effects resides in the presence of a significant number of “pain at injection sites” between phase designating the second dose of Placebo and the first dose of BNT162b2, for the Placebo who had been unblinded, and offered to receive the mRNA product. These events, designated in the ADAE file as occurring during the phase “After unblinding and before Vaccination 3”. As represented in the table below, some subjects wouldn’t even have received a second dose (R79) - and although 31 (60.8%) had this after-effect registered on the same day than their 3rd dose, it’s symptomatic of delays in registration of the third dose administration.

A manual review of the CRFs files highlighted that rather often, sites did not record any, or only some of the AEs that occurred in the CRFs. To detail but a few examples:
1055113980 - the CRF lists injection site soreness and chills, while the Adverse Events table (ADAE) additionally lists another event of chills, nausea, right axillary adenopathy and injection site pain (R81).

1140128282 - the CRF lists exercise induced asthma, while ADAE additionally lists bilateral pulmonary embolism, deep vein thrombosis, and right upper quadrant abdominal pain, along with a deleted AESPID #2. As there is only AESPID #1 in the the CRF, this means Pfizer added an adverse event and deleted it before adding the other three events, which are not in the CRF.

1241134783 - the CRF lists headache, injection site pain, lymphadenopathy and a second event of headache, while ADAE contains three additional events of fever, headache and fatigue.

The CRF’s AE log does not report any AEs for 35 subjects84, and at least 73 CRFs are missing AEs that are present in the database85, making a minimum of 108 subjects affected by this anomaly in a sample of only 1028, which leads us to question if the reporting was solely made at the sponsors’ discretion - and further, if the CRF files could have been altered. It constitutes at least a major violation of GCPs, and if it is confirmed that this isn’t human error but that CRFs have been altered, an infringement to several US86 & International regulations87.
COVID symptoms reported by the subjects
It seems necessary, to reach an objective representation of the capacity of the BNT162b2 to prevent COVID-like symptoms, to consider that if a subject reported a fever, no matter if Pfizer decided to classify it as “Adverse effects” or “COVID symptoms” - the fact remains that the subject reported a fever. We mapped the AEs and COVID symptoms into a single file88 that represented COVID-like symptoms, in order to represent the following unique new events reported by subjects during the “blinded” observation phase. We considered - as per protocol - reports of the same symptoms, per the same subjects, within 4 days89 - as a single symptom on the date of first occurrence (R90), no matter if the report of the condition affecting the subject was made via the reactogenicity dataset (FACE), the adverse events (ADAE) or the COVID symptoms (ADSYMPT). This means that if a subject reported a Fever and Cough on March 18, and another Fever on March 19, he would count only once, on March 18.

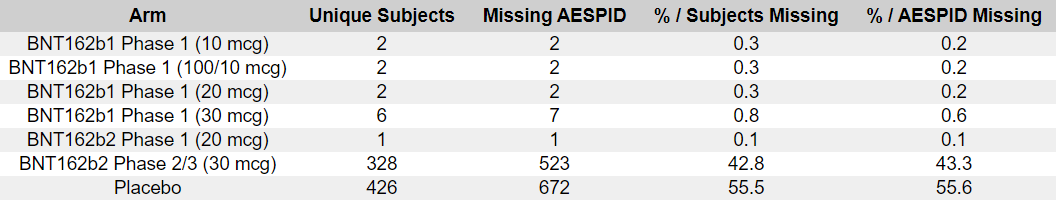
The unblinding wasn’t randomized
The Placebo recipients unblinding began as soon as the EUA was obtained, on December 14, 2020 (the FDA giving its authorization to it 3 days after the EUA on December 11, 2020). The subjects were unblinded non-randomly, prioritizing the elder subjects - and effectively terminating the Randomized Clinical Trial, which became an open-label study, with a very small and highly selective placebo group. The following chart represents the evolution of the cumulative unblinding rates for the 44 040 phase 2-3 subjects who had received their first dose prior December 14, 2020 (R91).
Forensic Analysis of the deaths during the trial
A peer reviewed paper92, “Forensic analysis of the 38 subject deaths in the 6-Month Interim Report of the Pfizer/BioNTech BNT162b2 mRNA Vaccine Clinical Trial” by Michels et al., found that Subject # 10841470, a 65-year-old obese Hispanic male with pulmonary fibrosis and hypertension was part of the placebo arm of a Pfizer/BioNTech COVID-19 vaccine trial. He received doses 1 and 2 of the placebo on September 30 and October 21, 2020. However, on December 23, 2020, he received a dose of the Moderna mRNA vaccine, which was a protocol deviation. He later developed COVID-19 symptoms, was hospitalized, and eventually died on January 11, 2021, due to multi-system organ failure. The case was misreported as a placebo death with COVID-19 as the secondary cause of death, when in fact, the subject should have been discontinued from the trial due to receiving a non-study COVID-19 vaccine.
The paper also notes,“a 3.7-fold increase in cardiac events in subjects who received the BNT162b2 vaccine versus the placebo. Of the 15 subjects who were Sudden Adult Deaths (SAD) or Found Dead (FD), 12 died of a cardiac event, 9 of whom were vaccinated.”
Delayed deaths recording at EUA time
Michels et al. highlights another major finding, further developed by Jeyanthi Kunadhasan in her letters exchanges with the Australian Therapeutic Goods Administration (TGA). These exchanges, sent and made public on behalf of the Australian Medical Professional Society (AMPS), are consisting in a first letter dated March 21, 202493, the reply of the TGA’s Health Products Regulation Group’s Professor dated March 27, 202494, and an expanded argument dated May 17, 202495 currently unanswered. They are attached to the current report. They provide another illustration of the diligence of the regulators for the matter of investigating critical irregularities - as the TGA interlocutor obviously fails to understand points clarified from the first letter.
A major anomaly lies in the delayed reporting of two deaths in the EUA figures communicated to the regulators, and to the world via the December NEJM study of Polack et al96. The Polack paper disclosed six deaths — two in the BNT162b2 arm and four in the placebo arm. Both the journal article and the EUA approval documentation showed the six deaths during the period of July 27, 2020, till November 14, 2020. The article shows that Pfizer-BioNTech had records showing eight deaths among the 11 deaths which had occurred at EUA time. The authors show that Pfizer had clear knowledge of four in the BNT162b2 arm and four in the placebo arm, and that Pfizer should have disclosed these to the FDA. The two undisclosed deaths indicated a cardiac event signal in the clinical trial’s BNT162b2 recipients. Subject 11141050 died on October 19th, 2020, well before the data cut-off date of November 14th, 2020. Documentation shows that the subject’s emergency contact notified the clinical site of the death on the date of death, as per protocol requirements. The protocol also required the clinical site to notify Pfizer, via its vaccine SAE form, within 24 hours of receiving a death notification. However, the clinical staff waited 37 days to enter this patient’s death into Pfizer’s records. Because of that delay, Pfizer did not submit this death as part of its EUA data, raising questions about the reasons for the delay and potential breaches of Good Clinical Practice.
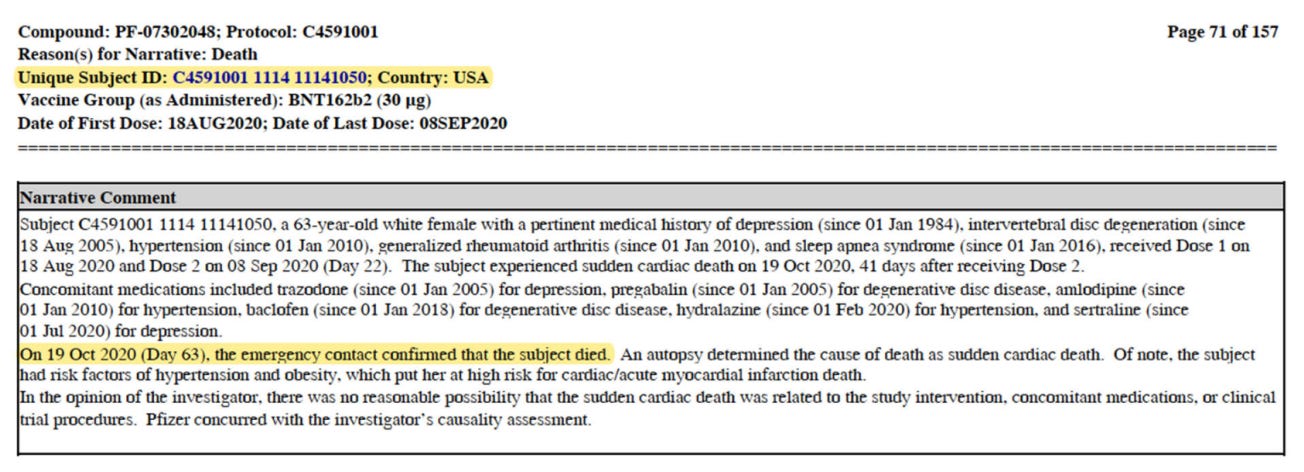
Per the autopsy, the patient died from ‘sudden cardiac death,’ with her known risk factors of hypertension and obesity putting her at high risk of cardiac-acute myocardial infarct. The clinical site staff entered the specific diagnosis of ‘sudden cardiac death’ into her notes on December 9th, 2020, the day before the Vaccine and Related Biologicals Products Advisory Committee (VRBPAC) meeting on December 10th, 2020, which suggests that this hidden death also had autopsy results available at the critical juncture of consideration of vaccine emergency use authorization.
To be eligible for inclusion in this clinical trial, participants had to be deemed healthy based on medical history, physical examination (if required), and the clinical judgement of the investigator. The protocol allowed healthy participants with pre-existing stable disease – defined as disease not requiring significant change in therapy or hospitalization for worsening disease during the six weeks before enrolment – to participate in the clinical trial. The blood pressure reading is absent of her publicly available case notes. Consequently, we can only assume the patient’s high blood pressure, from which she had suffered since January 1st, 2010, was well controlled when she was admitted to the trial. The patient weighed 74.1kg at a height of 165cm. Hence, her BMI of 27.2 placed her in the overweight category, not the obese one sustained, as referenced on point 74.
Subject 11201050 died on November 7th, 2020. Her husband reported her death to the clinical site on November 7th, 2020. Seventy-two days after receiving Dose 2 of the vaccine, she died in her sleep. No hospital visit or autopsy occurred. A coroner pronounced her death and listed the cause of death on her death certificate as cardiac arrest.

As no autopsy results were available, it remains unclear how the sponsors or the regulators concluded that this death could not be attributed to the treatment. Pfizer documented receiving notification of her death on November 7th, 2020, well before the data cutoff date of November 14th, 2020. The reasons for not disclosing this death from the vaccinated arm at the December 10th, 2020, VRBPAC meeting or in the Polack New England Journal of Medicine publication need clarification.
Confirming Michels et al. evaluation of massively under-reported deaths
Michels et al.’s notes that “the number of subject deaths was 17% of the expected number, based on age-adjusted US mortality. One possible explanation could lie in the 395 subjects that were“Lost to Follow-up”. Another explanation in these low deaths could lie in the offset of 1203 subjects in the investigators files highlighted on points 23 to 25 - and/or the 301 records missing (at least) highlighted on points 47 to 51.
No BNT162b2 recipient died outside of the United States (US) according to the reported figures. Deaths in the US are 19 subjects post BNT162b2, 12 subjects post Placebo, and 2 subjects first recipients of Placebo who had then received at least one dose of BNT162b2, for a total of 33 deaths in the US. 5 Placebos subjects died outside of the US.
We extracted the mortality data by state, year, month & 5 years age group brackets, for 2020 & 2021, from the CDC Wonder Underlying Causes of Death platform97 - and generated an export98, along with another export99 of the population100 yearly July 1st census data by states. This allowed us to proceed to a comparison of the mortality reported by the US trial sites ( and the mortality which could have been expected from a cohort of this age. Some healthy user bias would potentially have an impact - but the fact to observe a death from “dementia” allows to question the standards applied to the recruitment during the trial. This can’t account, in any case, for how significantly lower the mortality observed is than what would have been expected on a population of 33689 randomized individuals (R101) - with only 18% of the expected mortality being reported.

Process 1 & Process 2 - Differences overview
In the development of its COVID-19 vaccine candidate, BNT162b2 (commercially known as Comirnaty), Pfizer/BioNTech used two distinct manufacturing methods, which they referred to as “Process 1” and “Process 2.”
The research protocol102 for the clinical trial states: “The initial BNT162b2 was manufactured using ‘Process 1’; however, ‘Process 2’ was developed to support an increased scale of manufacture.” Additional evidence can be found in other regulatory documents from the US FDA, the Japanese Pharmaceuticals and Medical Devices Agency (PMDA), the TGA and the European Medicines Agency (EMA). To take one example, the EMA Product Assessment Report (PAR) for Comirnaty dated February 19, 2021, states: “Two active substance processes have been used during the development history; Process 1 (clinical trial material) and Process 2 (commercial process)”103.
In addition to the statement from the EMA PAR cited on point 83, which indicates that Process 1 was used for clinical trial doses while Process 2 was used for commercial doses, a corroborating statement comes from Japan’s PMDA in their “Report on Special Approval for Emergency” dated February 8, 2021: “The active substance used for nonclinical and clinical studies are manufactured by Process 1, and the active substance in the proposed commercial formulation by Process 2”104.
Amendment 7 to the protocol dated Oct. 6105 states: “Added an additional exploratory objective to describe safety and immunogenicity in participants 16 to 55 years of age vaccinated with study intervention produced by manufacturing ‘Process 1’ or ‘Process 2’”. This amendment is elaborated in section 6.1.1: “The scale of the BNT162b2 manufacturing has been increased to support future supply. BNT162b2 generated using the manufacturing process supporting an increased supply (‘Process 2’) will be administered to approximately 250 participants 16 to 55 years of age, per lot, in the study. The safety and immunogenicity of prophylactic BNT162b2 in individuals 16 to 55 years of age vaccinated with material generated using the existing manufacturing process ‘Process 1,’ and with material from lots generated using the manufacturing process supporting increased supply, ‘Process 2,’ will be described”106.
The clinical trial protocol cited in paragraph 65 summarizes three key differences between Process 1 and 2: “the process changes relate to the method of production for the DNA template that RNA drug substance is transcribed from, and the RNA drug substance purification method. The BNT162b2 drug product is then produced using a scaled-up LNP manufacturing process”107.
The differences between processes 1 and 2 are not trivial. A document obtained from the TGA108 details some of the key changes performed between “Process 1” and “Process 2”, although many details have been redacted. In Process 1, the DNA template for mRNA transcription was produced via PCR-amplification; Process 2 uses linearized plasmid DNA cultured in E. coli bacteria. Linearized Plasmid DNA involves the use of plasmid DNA, which is a small, circular, double-stranded DNA molecule typically found in bacteria. Additional (partially redacted) details described changes to the in-vitro transcription reaction volume, the batch scale, and the purification procedure (Magnetic Beads in Process 1; Proteinase K treatment to digest proteins and release associated nucleic acids followed by Tangential Flow Filtration in Process 2).
Identifying Process 2 lots
In some of the regulatory documents, Process 1 is sometimes referred to as a ‘classical’ process and the resulting product are referred to as the ‘clinical supply.’ Process 2 is sometimes referred to as the ‘upscale’ process and the resulting products are referred to as ‘emergency’ or ‘commercial’ supply. This is evident in a document made public by the TGA. It is FOI 3659 document 4, titled “BNT162b2 (PF-07302048) Comparability Report for PPQ Drug Product Lots”109, which lists the lots distributed, their use and their corresponding production processes.

The comparability report distinguishes clinical, emergency and commercial supply lots as follows: “The drug product manufacturing process has evolved from clinical supply to emergency supply and finally to commercial supply, undergoing transfer to different manufacturing sites and process scale-up. Clinical supply was initially produced at Polymun, Austria (“classical” process). In order to improve the mass throughput of the LNP process and increase the batch size, the process was scaled up at Polymun (“upscale” process) followed by transport of the bulk drug product (fully formulated LNPs prior to sterile filtration) to Pfizer Puurs, Belgium for fill/finish operations (for the manufacture of “emergency supply” and commercial supply). This process has also been set-up at mibe (Dermapharm), Germany for manufacture of emergency supply material. Within this scope, the fill/finish operations at Puurs were initially conducted on the S2F2 line (for clinical supply) with transition to the WSL5 line for larger batch volumes (emergency use supply, and subsequently for commercial supply). For routine commercial production, the LNP production process has been fully transferred to Pfizer, Puurs, Belgium with fill/finish operations on filling lines WSL5, FC2 and VC2 and to Pfizer, Kalamazoo, US with fill/finish operations on filling lines L8 and L18 (for the manufacture of “commercial supply”)”110.
We can identify which lots are Process 1 or Process 2 from the same document based on the lots listed in Table 1 on page 4 and in notes b and e to Table 3 on page 11. Additional information on the process used to manufacture lots is from a document released under FOI by the FDA111. This document is titled “125742_S2_M3_32p5_batch_analyses.pdf.” - Table 3.2.P.5.4-1 “Summary of BNT162b2 Drug Product Lots” on page 4 of the document contains the relevant information. Both of these documents will be referred to below.
Establishing Which Process 2 Lots Were Used in Clinical Trial C4591001
A document released by the FDA details which lots had been shipped to which clinical research site at approximately 6 months into the trial. It is dated March 17, 2021 and titled “125742_S1_M5_5351_c4591001-interim-mth6-patient-batches.pdf”112. The following unique lots numbers are listed in that document: BCV10320-A, BCV10420-A, BCV20420-A, BCV40420-A, BCV40620-A, BCV40620-B, BCV40620-C, BCV40620-D, BCV40720-A, BCV40720-B, BCV40720-C, ED3938, EE3813, EE8493Z, and EJ0553Z. It is not clear what the ‘Z’ at the end of lots EE8493 and EJ0553 refer to. In other documents these lots are listed without a Z suffix. It may have been used to designate that those lots used in the trial were from Process 2.
Based on the tables from TGA and FDA documents that were referred to in points 88 and 90 above, all lots starting with ‘BCV’ are Process 1 lots. Lot ED3983 is listed in Table 3.2.P.5.4-1 of the FDA batch analysis document cited in point 90 as a “clinical” lot and is marked with note a, which is defined on page 13 as “equivalent to BCV40720-P,” which was established as a process 1 lot on point 91. Lot EE3813 is listed as a Process 1 clinical lot in both tables.
Two Process 2 lots are identified in the 6-month patient batches document cited on point 91: EE8493 and EJ0553. Lot EE8493 is listed as a Process 2 lot in Table 1 of the TGA’s “Comparability Report for PPQ Drug Product Lots” document cited on point 90 above (see column marked “DS Process and Site of Manufacture.”). Lot EJ0553 is listed in the TGA comparability report in note ‘d’ of Table 3 on page 11. Note ‘d’ in Table 3 is attached to the column “Historical Range: Emergency Supply,” which indicates that EJ0553 is an emergency supply lot, which was another term for Process 2. Note ‘d’ further specifies that the manufacturing location of lot EJ0553 was “Polymun/Puurs WSL5”, which, based on the description from the document quoted on point 88, provides additional evidence that EJ0553 was an emergency supply lot and not a clinical lot, because every lot in Table 1 with the “DP Site of Manufacture” listed as “Polymun/Puurs WSL5” is a Process 2 lot. This is wholly consistent with the description of the evolution of the manufacturing process on page 3 as quoted in paragraph 70 above. Many of the other lots in note ‘d’ are listed in Table 1 on pages 4-5 of the TGA comparability report and are all marked as Process 2, and none of the lots listed in note ‘d’ under Table 3 are listed in Table 1 as a clinical (Process 1) lot. In addition, all clinical lots are listed separately in note ‘b’ of Table 3 on page 11. Lot EJ0553 is listed in the FDA “Batch Analysis” document cited on point 90 as both an emergency supply and clinical lot. Given that lot EE8493 is also listed as both an emergency supply and clinical lot, this indicates that both EE8493 and EJ0553 were used in some capacity in the clinical trial and were also part of the emergency supply.
In addition, almost all adverse events reported113 in the United States Centers for Disease Control’s Vaccine Adverse Events Reporting System (VAERS114) for lots EE8493 and EJ0553 have a date of vaccination after Comirnaty had been authorized for temporary use in the UK. None of the other lots listed as having been supplied to the research sites have adverse event reports in VAERS115. This provides additional evidence that EE8493 and EJ0553 were process 2 formulations, as they were distributed and administered commercially post authorization.
Evidence Lot EE8493 Was Used for Planned Comparison of Processes 1 and 2
There are two ways to identify that lot EE8493 was used as part of the planned comparison study between Processes 1 and 2 as described in the clinical trial protocol cited on point 85. The first is by identifying which Process 2 lots were shipped to which clinical trial research sites and count how many treatment subjects were enrolled from the beginning of the comparison study (see points 98-99 below). The second is to show that the randomization numbers of a subset of participants was distinct from all the others (see point 100 below). These independent methods converge on the same result: only 252 subjects given vaccine doses from lot EE8493 were eventually enrolled as part of the planned comparison.
In a reply to FOI request 23/510, the United Kingdom’s Medicines & Healthcare products Regulatory Agency (MHRA) attested that “the first clinical batch which contained process 2 drug substance was dosed 19th October 2020 in US”116.

The document from the FDA cited on point 91 lists which lots had been distributed to which study sites as of March 17, 2021. A similar document117 lists which lots were supplied to each study site as of November 19, 2020. In that document, the only Process 2 lot listed is EE8493 (note: lot EJ0553 is not listed). Therefore, it can be inferred that EE8493 was the only Process 2 lot that had been distributed to the clinical study sites when the comparative study of processes 1 vs. 2 began on October 19, 2020. It was supplied to sites 1133, 1135, 1146 and 1170 according to both the interim and 6-month patient batch documents.
Analysis of the subjects who received their first dose on or after October 19, 2020 (R118) shows that at those four sites, there were a total of 252 subjects administered the vaccine and 250 administered the placebo119. All of the 252 subjects in the treatment arm were between the ages of 16 and 55. This is consistent with the description of the planned study in the protocol to administer Process 2 doses “to approximately 250 participants 16 to 55 years of age” as detailed in paragraph 2. (See Appendix A for a list of the 252 subjects recruited to these sites during this time period.) This analysis can be confirmed by visual inspection of the BLA randomization file communicated by the sponsors to the FDA120.
Lot EJ0553 was shipped to site 1007. The 17 treatment recipients at site 1007 after October 19, 2020 do not have randomization numbers that indicate inclusion in the comparative study of Processes 1 and 2, as detailed in paragraph 100. In addition, there are only 17 treatment subjects who could have potentially received doses from lot EJ0553, not the approximately 250 as planned in the protocol cited in paragraph 2. Therefore, we can conclude that these 17 subjects were not part of the planned study to compare processes 1 and 2. Given that lot EJ0553 was supplied to three other trial sites that did not enroll any new treatment subjects after November 14, 2020, it is likely that doses from this Process 2 lot were supplied to those four sites to administer to placebo subjects who were unblinded and offered to be vaccinated subsequent to the FDA’s issuance of Emergency Use Authorization on December 11, 2020.
The dataset and documents used for this analysis provide a second means for identifying the subjects involved in the comparison study - discovered by the same authors who authored Michels et al. (cited on points 71 to 79): the subject randomization number (R121). Every subject in the trial falls between the numbers 1081 and 274318, except for a small subset of 502 subjects who have a unique range of randomization numbers between 400002 and 401509. Of these, 252 are the same treatment subjects identified on point 98, at the four sites that received Process 2 lots prior to Nov. 19, 2020. The others 250 are Placebo recipients, the same than those at the four sites identified on point 97 (R122). This shows independent corroboration of the conclusion from paragraph 80 that these 252 treatment subjects from sites that received lot EE8493 were the only ones who were intended for the comparison between processes 1 and 2.
The comparison of process 1 vs process 2 recruitment has been represented on a weekly chart.

Evidence The Planned Comparison of Processes 1 and 2 Was Never Conducted
In its response to FOI request 23/510, cited in paragraph 96 and attached to this report, the MHRA states that the data from the study were expected in February, 2021 (page 2). However, the planned comparison from the protocol was “removed and documented in Protocol amendment 20 in September 2022 due to the extensive usage of vaccines manufactured via ‘Process 2’. Thus, this process comparison was not conducted as part of the formal documentation” (page 5). At least as far as the MHRA is concerned, the comparison study was never conducted.
Evidence the two products aren’t perfectly similar : higher rates of adverse effects among Process 2 recipients
We modeled the impact of being a recipient of Process 2 compared to other variables on the number of adverse effects registered by subjects in the USA, 55 or under, using a Poisson regression, and found that it increases the count of adverse events by 264.33%; having a highly significant impact on the adverse effects (6.59 times larger effect than “comorbidity”); although less significant than the impact of the sex or obesity (R123).
Comparing exclusively the 95 non-obese male recipients male recipients in the USA, Process 2 results in a 211.77% increase, with an impact 4.84 times larger than comorbidity. The evaluation is similar (214.49% increase) using a negative binomial regression.
Evidence of Differences in Lymphadenopathy for Process 1 versus Process 2 Lots and Inaccurate Statements by the MHRA
Lymphadenopathy is the medical term for swollen lymph nodes. Differences in the reported rate of lymphadenopathy between the first two vaccine doses in the clinical trial (C4591001) and the rate of lymphadenopathy from the third (booster) dose are indicative of the different adverse event profiles of process 1 versus process 2 vaccine product. The reason is that the first two doses in the clinical trial were overwhelmingly from vaccine doses made with process 1, whereas booster doses given many months later would have been manufactured using the process 2 for commercial supply.
The “MHRA’s Public Assessment Report for the Authorisation for Temporary Supply” of Pfizer/BioNTech’s COVID-19 mRNA Vaccine BNT162b2 describes the rate of lymphadenopathy based on the preliminary report from clinical trial C4591001124. Although the public assessment report (MHRA’s PAR) was updated for adolescent vaccination in June, 2021, the cover page states that it “summarises the initial assessment at the time of approval in December 2020. The text in the original report remains unchanged.”
The lymphadenopathy rates from clinical trial C4591001 are listed on page 42 of the MHRA’s PAR as 0.3% and classed on page 50 as “uncommon,” defined as occurring greater than 0.1% of the time but less than 1%.
In the MHRA’s June, 2023, document titled “Information for Healthcare Professionals on COVID-19 Vaccine Pfizer/BioNTech (Regulation 174)”, the lymphadenopathy rate is given in Table 1125. It is listed as uncommon. But note (a) at the bottom of the table states: “A higher frequency of lymphadenopathy (5.2% vs 0.4%) was observed in participants receiving a booster dose (third dose) compared to participants receiving 2 doses.” A rate of 5.2% would move the lymphadenopathy from the ‘uncommon’ classification to ‘common.’ It represents a greater than 13-fold increase in the rate of lymphadenopathy126.
One possible explanation for this increase is that the booster dose provokes a stronger immune reaction, which leads to a higher rate of lymphadenopathy. However, careful and highly accurate imaging studies of lymphadenopathy performed by Israeli researchers at Tel Aviv University’s Medical School using PET-CT scans concluded that “the overall incidence of any-grade VAHL [vaccine-associated hypermetabolic lymphadenopathy] following the third COVID-19 vaccine dose is basically similar to that reported following the first and second COVID-19 vaccine doses. However, VAHL cases following the third dose were found to have shorter duration and low uptake intensity after the first 5 days from vaccination”127. This means not only that lymphadenopathy following booster vaccination was not worse than the first two doses, but in some ways it was even less severe.
The results of the Israeli research are not consistent with the 13-fold increase in the rate of lymphadenopathy reported by MHRA between the first two doses and the third dose. This Israeli research was not conducted as part of clinical trial C4591001. It was based on individuals who received commercial doses supplied to Israel after the vaccine rollout, meaning doses made using process 2.
On this basis, it can be concluded with very high confidence that the increase in the rate of lymphadenopathy reported by the MHRA is due to the switch from process 1 doses used in the clinical trial to process 2 doses used later for booster doses.
Although the source of the 5.2% rate of lymphadenopathy is not given, in light of the timing at which booster doses were first administered in the second half of 2021, they would had to have come from booster doses made using process 2. An increased rate of lymphadenopathy in booster doses is also supported by the Pfizer/BioNTech clinical trial C4591031 on the first booster (3rd dose), which reported a lymphadenopathy rate of 2.7%128. This represents an increase of over 6 times from doses 1 and 2 and is also not consistent with the finding from the Israeli research cited above (see fn. 12) showing no difference in lymphadenopathy between the first booster and earlier doses.
The Cut-off Dates weren’t “Data Cut-off”
While traditional trials have a date of cut-off which corresponds to the chronological end of the study, here the trial continued both after the EUA and the BLA applications. Practically, while the BLA data submitted documents indicating a cut-off date of March 13, 2021, the CRF kept being edited up to April 1st, 2021, as visual inspection of any CRF file reveals - see for example point 65. This makes reproduction of the BLA application - such as the exact COVID cases communicated, nearly impossible without full access to the audit trail. A vast quantity of the adverse effects “re-qualified to COVID symptoms” are concerned by these “last minute edits”. To provide but a few examples of ambiguities related to the data cutoff, to be found among the 1028 CRF files:
12231058129 - the adverse events of fatigue and injection site pain occurring on March 2nd & 3th, 2021, were entered on March 23, 2021, and are present in the database (R130).

12261769131 - the subject has a COVID visit on March 18, 2021 entered on March 19, 2021 that is not present in the database.
12312420132 - COVID visit A from March 12 to March 15, 2021, entered March 16, 2021, and COVID visit B, 26.3. - ongoing, entered March 29, 2021, with a positive local swab on page 235, are both not present in the database.
12313610133 - the SAE of atrial flutter, which occurred on February 19, 2021., was entered on March 20, 2021, with the site having sent the initial safety database on March 19, 2021, page 181, and is present in the database.

Exploitation of the Medical History Loophole
The Study Protocol134 specified major loopholes in the recording of the adverse events, practically meaning that preexisting conditions or conditions parts of the medical history of the subject wouldn’t be registered depending on the appreciation, by the principal investigator, of the “related nature” of the condition.

This protocol loophole, whose full characterization is a tedious work which isn’t the subject of this current report, has allowed the dissimulation of considerable “worsening of conditions”. For example, subject 10791044135 sees a “worsening of osteoarthritis”, resulting in his two knees being replaced, or 11171146’s aortic aneurysm being deleted from the AE log due to the PI’s assessment.136

Evidence the two products aren’t perfectly similar: elevated rates of Menorrhagia in post-marketing studies
Posterior studies found high rates of Menorrhagia ; for example 12+%137 in a study on female students and staff at a university in Saudi Arabia. A meta-analysis138, by Al Kadri, found a pooled prevalence of 24.24 % (95 % CI: 12.8–35.6 %).
By opposition, only 4 cases of Menorrhagia had been reported as Adverse events during the trial (R139) - 2 among the BNT162b2 recipients (0.065% of the 3087 women 16 to 39).
Inspecting the Medical History records (R140), we found 131 BNT (1.77%) recipient female subjects 16 to 59, with a past medical history of Menorrhagia - for all of whom the event status was set to “LAST SUBJECT ENCOUNTER”. The rate increases to 1.93% (143 subjects) if we include other conditions which could be considered by the PI has related to Menorrhagia (Dysfunctional uterine bleeding, Ovarian haemorrhage). This remains very far from the post-study rates, and would lead us to conclude that the very high rates of Menorrhagia in the post-marketing studies are another consequence of the switch to Process 2.
Anomalous patterns in PCR Testing
There were a total of 110 days of the study from July 27, 2020 - first randomization, to the November 14, 2020 data cut-off, last data point included in the submission to the VRBPAC and the Polack paper in the New England Journal of Medicine. During this time period, following their first dose, 448 positive PCR tests were recorded in the Placebo arm, and 163 positive tests in the BNT162b2 arm (R141).

Restricting, per protocol, to the tests recorded with symptoms within 4 days, brings the totals to 306 Placebo & 55 BNT162b2. The rate of BNT162b2 discarded by lack of symptoms (-66.3%) against the Placebo discarded (-31.7%) is of course highly statistically significant.

We highlighted on point 69 the impact of the re-qualifications of the COVID symptoms. It is therefore useful to proceed to a similar “symptomatic analysis” considering COVID symptoms logged, no matter the dataset sustained by the clinical trial staff. This angle brings the totals to 70 symptomatic BNT162b2 with positive PCR (+15) against 316 Placebo (+10), and doesn’t explain the imbalance. The accrual of symptomatic PCR tests for each arm is represented in the following chart.

We analyzed by country (R142), for USA & Argentina, the percentage of subjects reporting symptoms, resulting in a Central PCR test within 4 days, as per protocol. We smoothed the rendering on a rolling average of 3 days. The following chart exposes how, in the USA, each wave of symptoms reported is followed by a significant drop in the testing of both arms. Although it is possible that tests shortages would have occurred during high COVID waves, it is - in absence of adequate documentation - also possible that tests manipulations, such as delaying the recording of symptoms, of other forms of tests mitigation, would have occurred during these periods of drastic testing rate drops. This isn’t, in any case, the testing pattern we could expect in a trial aiming to provide reliable results.
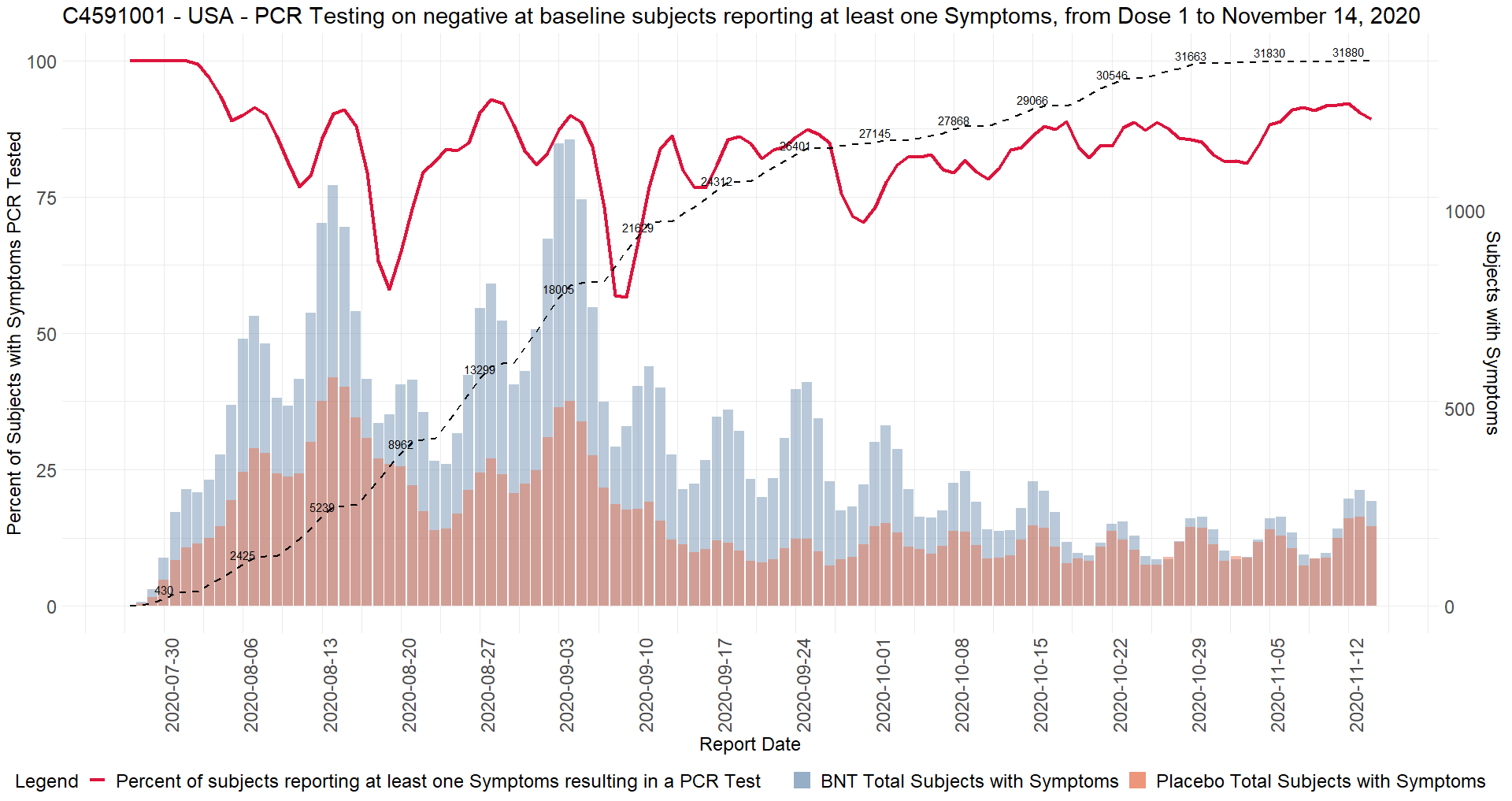
The same visible pattern, of drastic drop of the testing rate following the periods of intensive symptoms reporting, is visible - and also happens while symptoms reported are low, in Argentina (R143) - posing further questions on the events which impacted the testing of the subjects reporting symptoms.

Anomalies related to the PCR testing on subjects’ COVID visits
The randomized subjects in phase 3 triggered a total of 10,219 "symptomatic visits" - each "symptomatic visit" corresponding to a suspected COVID due to symptoms, but which may generate several physical visits by the subject to the site at several days' intervals (R144).
According to the protocol, samples taken during symptomatic visits were to be tested locally and "centrally", at the Pfizer Pearl River laboratory, New York, USA. The central laboratory tests were the preferred method for a COVID case to be confirmed145, although depending on protocol variations, local tests approved by the FDA could be used146. A total of 9474 central tests and 5489 local tests were recorded at the cut-off date of March 13, 2021.
87.2% of the 4421 symptomatic visits made by subjects in the BNT162b2 group resulted in PCR tests being sent to the central laboratory, compared with 88.3% of the 5798 visits made by subjects in the placebo group. This minor difference was not significant.
In contrast, at the same date, only 43.5% of the 4421 symptomatic visits made by subjects in the BNT162b2 group had resulted in local PCR tests, compared with 50.8% of the 5798 visits made by subjects in the placebo group. This difference is highly significant and demonstrates that a significant difference in treatment between the groups, by the teams in charge, occurred at certain sites.

It is also important to observe the treatment of subjects for PCR tests on symptomatic visits at the end of the Emergency Use Authorization (EUA) in the United States on November 14, 2020. By this date, 5415 symptomatic visits had been recorded. Only 65.1% of BNT162b2 & 65.5% of Placebo patients had had their symptomatic visits result in central testing.
Although less statistically significant, the difference in treatment for local tests persists. This difference in treatment, which was evident before the emergency marketing authorization was obtained, has not yet been explained.

Analyzing symptomatic visits resulting in central PCR tests within the protocol window (R147), the number of tests carried out within the time limit drops to 54.2% for BNT162b2 and 56.2% for Placebos. The proportion of local tests carried out within the protocol window also falls, while continuing to show a significant difference in treatment between the groups, with 29.2% of tests done for the BNT162b2 and 33.3% for the Placebos (p-value 8.103e-06).
Analysis of the local tests by sites shows the infringements to the blind are located in the United States
Returning to the perspective envisaged in point 127 (March 13, 2021 by COVID visits - otherwise said as the BLA data was analyzed), we can focus on these anomalies by test site in order to identify abnormal sites (R148). We performed a Fisher's exact test on sites with at least 50 symptomatic visits in total in order to identify significantly unbalanced sites - systematically under-testing the BNT162b2 group compared to the Placebo group.
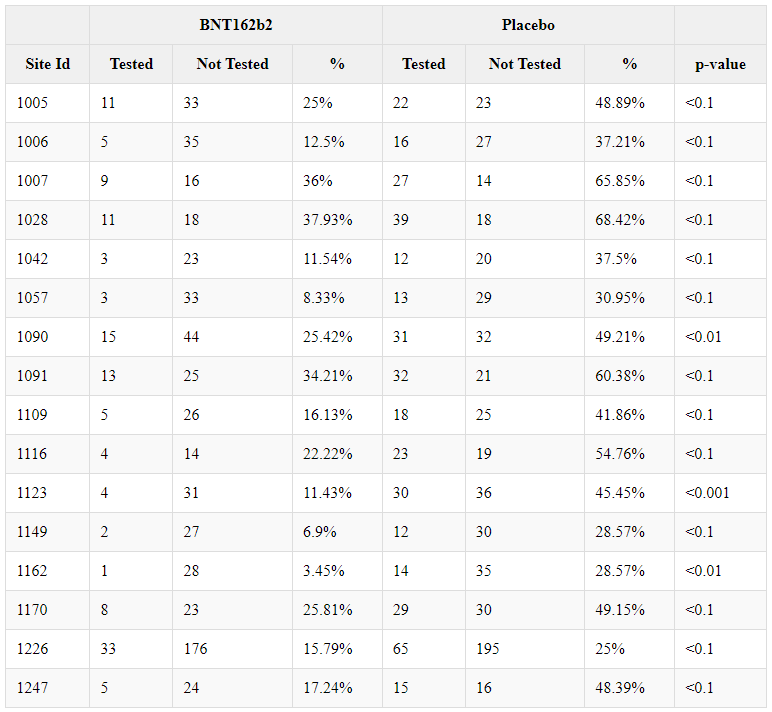
To evaluate if less frequent, varied or severe presentations of symptoms could explain the offset, we measured the difference between symptoms reported by subjects and visits, on these sites, using a t-test (t = -4.729, p-value = 2.451e-06). The mean of symptoms reported by subjects on their visits was 2.49 for the BNT against 2.89 for the Placebos, would indicate that the BNT subjects reported significantly less symptoms than the Placebo.
However, given the questionable categorization of symptoms from COVID visits to adverse events we highlighted via Augusto Roux’s testimony on point 46, and the wide scale of the phenomenon shown on points 61 to 66, it is necessary to observe that this effect reverses if we consider the COVID-like symptoms reported across all datasets. The following chart represents the percentage of symptoms reported by subjects, with a 7 days rolling average, in both arms, which resulted in being qualified as a COVID symptom and associated to a COVID visit in the adequate dataset. It illustrates that the proportion of symptoms reported by the BNT ending in being qualified as symptomatic visits remained consistently lower, up to the EUA & unblinding.
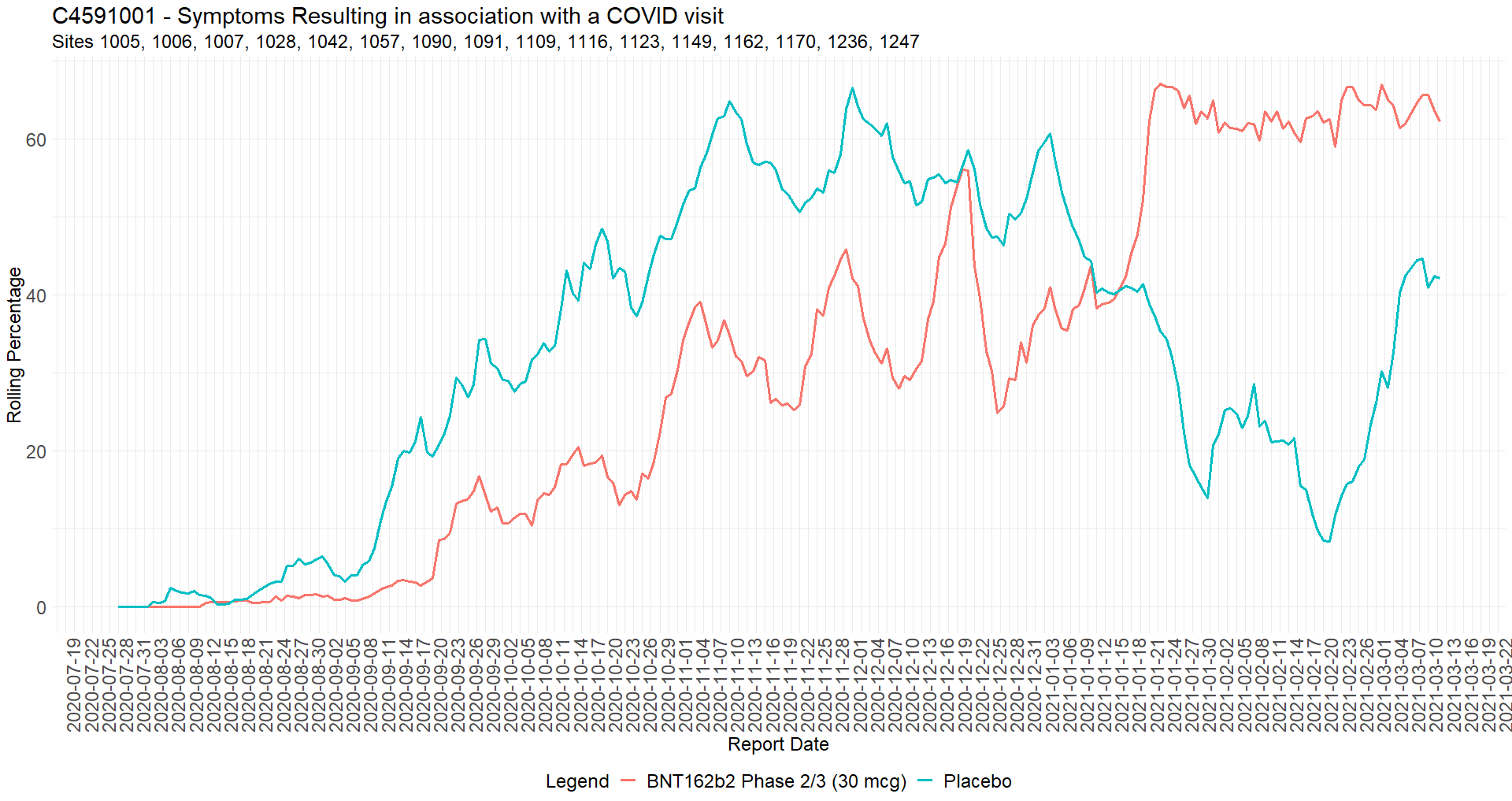
Recapitulation of the major anomalies at sites scale
The major anomalies at “site scale” can be categorized between those impacting the blind:
anomalies to deviations, Point 33
anomalies in the PCR rates between arms, Point 131
and those involving serious concerns that subjects may have been removed from the records:
anomalies in the randomization numbers communicated, Point 25
anomalies in the identification numbers of the subjects, Point 48
The following tables are summarizing the impacted sites ; leaving only 17 sites over the 153 listed to be “spared” by major anomalies.
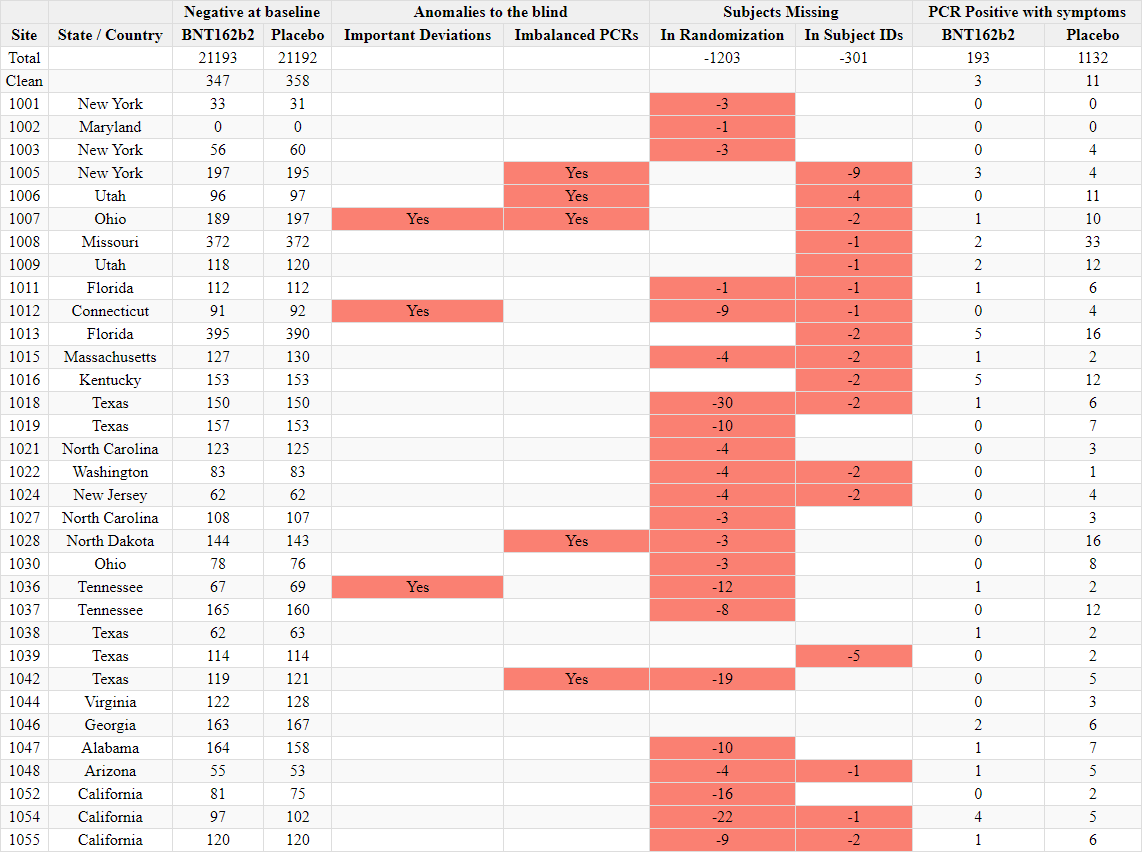




FDA.gov - Ramachandra Naik, Ph.D et al. - Emergency Use Authorization (EUA) for an Unapproved Product Review Memorandum - https://www.fda.gov/media/144416/download
STATNews.com - Peter Doshi and Matthew Herder - Did the FDA understaff its review of the Pfizer/BioNTech vaccine? - https://www.statnews.com/2020/12/17/did-the-fda-understaff-its-review-of-the-pfizer-biontech-vaccine
PHMPT.org - UNITED STATES DISTRICT COURT FOR THE NORTHERN DISTRICT OF TEXAS FORT WORTH DIVISION - https://phmpt.org/wp-content/uploads/2022/01/ORDER_2022_01_06.pdf
PHMPT.org - Public Health and Medical Professionals for Transparency -
R-4.4.0 for Windows - https://cran.r-project.org/bin/windows/base
R Script - download_full_prod.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/download_full_prod.R
R Script - extract_full_prod.R -
https://github.com/OpenVaet/pfizer_docs_R/blob/main/extract_full_prod.R
PHMPT.org - Pfizer 16+ Documents - https://phmpt.org/pfizer-16-plus-documents
pfizer.com - Pfizer and BioNTech to Submit Emergency Use Authorization Request Today to the U.S. FDA for COVID-19 Vaccine - https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-biontech-conclude-phase-3-study-covid-19-vaccine
FDA.gov - Susan Wollersheim, MD and Ann Schwartz, MD - BLA Clinical Review Memorandum - https://www.fda.gov/media/152256/download
R Script - extract_full_prod.R - screening_dates_range.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/screening_dates_range.R
PHMPT.org - Clinical Study Data Reviewer’s Guide - https://phmpt.org/wp-content/uploads/2022/05/125742_S1_M5_c4591001-S-csdrg.pdf, Page 24
PHMPT.org - 5.2-listing-of-clinical-sites-and-cvs-pages-1-41.pdf - https://phmpt.org/wp-content/uploads/2021/11/5.2-listing-of-clinical-sites-and-cvs-pages-1-41.pdf
R Script - screenings_by_weeks.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/screenings_by_weeks.R
Documentation - trial_site_data.json - https://github.com/OpenVaet/pfizer_docs_R/blob/main/trial_site_data.json
R Script - subjects_by_arms_and_sites.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/subjects_by_arms_and_sites.R
R Script - generate_screening_map.R, Scale: 1 subject / 100,000km² - https://github.com/OpenVaet/pfizer_docs_R/blob/main/generate_screening_map.R
PHMPT.org - C4591001 Investigational BNT162 Vaccine Program PF-07302048 - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-oversight-committees.pdf, Page 42
FDA.gov - Susan Wollersheim, MD and Ann Schwartz, MD - BLA Clinical Review Memorandum - https://www.fda.gov/media/152256/download, Page 5
The New England Journal of Medicine - Edward E. Walsh, M.D et al. - Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates - https://www.nejm.org/doi/10.1056/NEJMoa2027906
Nature - Mark J. Mulligan et al., Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults - https://www.nature.com/articles/s41586-020-2639-4
R Script - screening_dates_range_phase1.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/screening_dates_range_phase1.R
PHMPT.org - Protocol C4591001 Protocol Amendment 9, 29 October 2020 - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 87
PHMPT.org - Statistical Analysis Plan - https://phmpt.org/wp-content/uploads/2022/07/125742_S1_M5_5351_c4591001-fa-interim-sap.pdf, Pages 37 & 38
R Script - subjects_by_arms_and_sites_phase1.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/subjects_by_arms_and_sites_phase1.R
R Script - generate_screening_map_phase1.R, Scale: 1 subject / 100,000km² - https://github.com/OpenVaet/pfizer_docs_R/blob/main/generate_screening_map_phase1.R
PHMPT.org - 125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, Pages 1974, 1782, 1695, 32
PHMPT.org - 125742_S1_M5_5351_c4591001-fa-interim-iec-irb-consent-form.pdf, CONSENT TO TAKE PART IN STUDY - https://phmpt.org/wp-content/uploads/2022/04/125742_S1_M5_5351_c4591001-fa-interim-iec-irb-consent-form.pdf, Page 87
R Script - phase_1_adva.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/phase_1_adva.R
The New England Journal of Medicine - Edward E. Walsh, M.D et al. - https://www.nejm.org/doi/pdf/10.1056/NEJMoa2027906 - Page 10
Documentation - phase_1_different_adva_results.csv - https://github.com/OpenVaet/pfizer_docs_R/blob/main/phase_1_different_adva_results.csv
PHMPT.org - Analysis Data Reviewer Guide, 125742_S1_M5_c4591001-A-adrg.pdf - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_c4591001-A-adrg.pdf, Page 33
PHMPT.org - 2.7.1 Summary of Biopharmaceutic Studies and Associated Analytical Methods https://phmpt.org/wp-content/uploads/2022/06/125742_S1_M2_summary-biopharm.pdf, Page 3
PHMPT.org - Qualification of the SARS-CoV-2 mNeonGreen Virus Microneutralization Assay https://phmpt.org/wp-content/uploads/2023/04/125742_S1_M5_5314_vr-mqr-10214.pdf
PHMPT.org - Method Validation of the SARS-CoV-2 mNeonGreen Virus Microneutralization Assay https://phmpt.org/wp-content/uploads/2023/04/125742_S1_M5_5314_vr-mvr-10083.pdf
PHMPT.org - 125742_S1_M5_5351_c4591001-fa-interim-iec-irb-consent-form.pdf, CONSENT TO TAKE PART IN STUDY - https://phmpt.org/wp-content/uploads/2022/04/125742_S1_M5_5351_c4591001-fa-interim-iec-irb-consent-form.pdf, Page 67
PHMPT.org - 125742_S1_M5_5351_c4591001-fa-interim-investigators.pdf, 16.1.4.1 LIST OF INVESTIGATORS AND NUMBER OF SITES AND SUBJECTS BY THE COUNTRY - https://phmpt.org/wp-content/uploads/2023/10/125742_S1_M5_5351_c4591001-fa-interim-investigators.pdf
PHMPT.org - 125742_S1_M5_5351_c4591001-interim-mth6-investigators.pdf, 16.1.4.1 LIST OF INVESTIGATORS AND NUMBER OF SITES AND SUBJECTS BY THE COUNTRY - https://phmpt.org/wp-content/uploads/2023/10/125742_S1_M5_5351_c4591001-interim-mth6-investigators.pdf
R Script - investigators_screening_rando.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/investigators_screening_rando.R
Documentation - offset_randomization_between_fa_m6.csv - https://github.com/OpenVaet/pfizer_docs_R/blob/main/offset_randomization_between_fa_m6.csv
PHMPT.org - Analysis Data Reviewer Guide, 125742_S1_M5_c4591001-A-adrg.pdf - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_c4591001-A-adrg.pdf, Page 20
FDA.gov - Susan Wollersheim, MD and Ann Schwartz, MD - BLA Clinical Review Memorandum - https://www.fda.gov/media/152256/download, Page 7
The New England Journal of Medicine - Stephen J. Thomas, M.D. et al. - Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months - https://www.nejm.org/doi/10.1056/NEJMoa2110345 - “Between July 27, 2020, and October 29, 2020, a total of 45,441 participants 16 years of age or older underwent screening, and 44,165 underwent randomization at 152 sites”.
R Script - reproduce_rando_march_13.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/reproduce_rando_march_13.R
Documentation - phase_3_randomized_pop_stats.csv - https://github.com/OpenVaet/pfizer_docs_R/blob/main/phase_3_randomized_pop_stats.csv
PHMPT.org - Protocol C4591001 Protocol Amendment 9, 29 October 2020 - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 57
R Script - format_deviations.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/format_deviations.R
PHMPT.org - Protocol C4591001 Protocol Amendment 9, 29 October 2020 - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 64
FDA.gov - Ramachandra Naik, Ph.D et al. - Emergency Use Authorization (EUA) for an Unapproved Product Review Memorandum - https://www.fda.gov/media/144416/download, Page 14
PHMPT.org - Analysis Data Reviewer Guide, 125742_S1_M5_c4591001-A-adrg.pdf - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_c4591001-A-adrg.pdf, Pages 66 & 67
PHMPT.org - 125742_S1_M5_CRF_c4591001-1016-10161289.pdf - https://phmpt.org/wp-content/uploads/2023/05/125742_S1_M5_CRF_c4591001-1016-10161289.pdf, Page 270
Ema.europa.eu - ICH guideline for good clinical practice E6(R2) - Step 5 - https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-good-clinical-practice-e6r2-step-5_en.pdf, Section 4.11 - Safety Reporting, Section 4.9 - Records and Reports, Section 5.17 - Monitoring, Section 8 - Essential Documents for the Conduct of a Clinical Trial
PHMPT.org - 125742_S1_M5_CRF_c4591001-1231-12315632.pdf - https://phmpt.org/wp-content/uploads/2023/06/125742_S1_M5_CRF_c4591001-1231-12315632.pdf, Page 301 & 314
TheBMJ - Paul D Thacker - Covid-19: Researcher blows the whistle on data integrity issues in Pfizer’s vaccine trial - https://www.bmj.com/content/bmj/375/bmj.n2635.full.pdf
davidhealy.org - Dr David Healy - Disappeared in Argentina - https://davidhealy.org/disappeared-in-argentina
IOS Press - David Healy, Augusto Germán Roux, Brianne Dressen - The coverage of medical injuries in company trial informed consent forms - https://content.iospress.com/articles/international-journal-of-risk-and-safety-in-medicine/jrs220043
R Script - subject_12312982_aes.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/subject_12312982_aes.R
R Script - verify_301.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/verify_301.R
PHMPT.org - Statistical Analysis Plan - https://phmpt.org/wp-content/uploads/2022/07/125742_S1_M5_5351_c4591001-fa-interim-sap.pdf, Pages 13 & 46
PHMPT.org - 125742_S1_M5_5351_c4591001-fa-interim-lab-measurements-sensitive.pdf, Table 16.2.8.2 - Listing of Subjects With First COVID-19 Occurrence From 7 Days After Dose 2 and Without Evidence of Infection Prior to 7 Days After Dose 2 – Evaluable Efficacy (7 Days) Population - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-lab-measurements-sensitive.pdf, Page 66 to 99
R Script - extract_official_EUA_cases.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/extract_official_EUA_cases.R
R Script - analyze_eua_official_cases.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/analyze_eua_official_cases.R
Youtube - Pfizer & The National Geographic - Mission Possible, Race for a Vaccine - https://youtu.be/jbZUZ9JYNBE, 00:30:13 to 00:31:17
Spectator, Australia - Jeyanthi Kunadhasan, MD - 170 patients that changed everything - https://www.spectator.com.au/2022/12/170-patients-that-changed-everything, archived at web.archive.org/web/20221208045723/https://www.spectator.com.au/2022/12/170-patients-that-changed-everything/
R Script - eua_cases_deviations.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/eua_cases_deviations.R
PHMPT.org - 125742_S1_M5_5351_c4591001-fa-interim-discontinued-patients.pdf, 16.2.1.4 Listing of Subjects Withdrawn From the Study – All Subjects - https://phmpt.org/wp-content/uploads/2022/06/125742_S1_M5_5351_c4591001-fa-interim-discontinued-patients.pdf, Page 78
Nature Communications - Antonio E. Muruato et al. - A high-throughput neutralizing antibody assay for COVID-19 diagnosis and vaccine evaluation - https://ncbi.nlm.nih.gov/pmc/articles/PMC7426916
MDPI - Ming Luo et al. - Nucleocapsid Structure of Negative Strand RNA Virus - https://www.mdpi.com/1999-4915/12/8/835
Oxford Academic - Michael D Swartz et al. - Antibody Duration After Infection From SARS-CoV-2 in the Texas Coronavirus Antibody Response Survey - https://academic.oup.com/jid/article/227/2/193/6581498
R Script - anayse_n_binding_pcr.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/anayse_n_binding_pcr.R
PHMPT.org - Annotated Study Book for Study Design: C4591001 - https://phmpt.org/wp-content/uploads/2022/05/125742_S1_M5_c4591001-S-acrf.pdf, Page 11
PHMPT.org - Clinical Study Data Reviewer’s Guide - https://phmpt.org/wp-content/uploads/2022/05/125742_S1_M5_c4591001-S-csdrg.pdf, Page 34
R Script - subjects_adverse_effects_deleted.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/subjects_adverse_effects_deleted.R
PHMPT.org - 125742_S1_M1_meeting-correspondence.pdf, 1.6.3 CORRESPONDENCE REGARDING MEETINGS - https://phmpt.org/wp-content/uploads/2023/10/125742_S1_M1_meeting-correspondence.pdf, Page 574
PHMPT.org - 125742_S1_M5_CRF_c4591001-1218-12181001.pdf - https://phmpt.org/wp-content/uploads/2023/05/125742_S1_M5_CRF_c4591001-1218-12181001.pdf, Page 401
PHMPT.org - 125742_S1_M5_CRF_c4591001-1087-10871286.pdf - https://phmpt.org/wp-content/uploads/2023/08/125742_S1_M5_CRF_c4591001-1087-10871286.pdf
R Script - aes_between_d2_d3.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/aes_between_d2_d3.R
PHMPT.org - CRFs for site 1055.pdf - https://phmpt.org/wp-content/uploads/2021/12/CRFs-for-site-1055.pdf, Pages 1125 to 1403
R Script - aes_crfs_missing.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/aes_crfs_missing.R
PHMPT.org - 125742_S1_M5_CRF_c4591001-1140-11401282.pdf - https://phmpt.org/wp-content/uploads/2023/08/125742_S1_M5_CRF_c4591001-1140-11401282.pdf
PHMPT.org - 125742_S1_M5_CRF_c4591001-1241-12411347.pdf - https://phmpt.org/wp-content/uploads/2023/08/125742_S1_M5_CRF_c4591001-1241-12411347.pdf
Subjects with no AEs in the CRF file, yet present in ADAE: 10081603, 10371141, 10371214, 10771137, 10811102, 10851018, 10901492, 10961181, 10961355, 11071196, 11101164, 11101165, 11171079, 11261244, 11281241, 11281267, 11331317, 11341327, 11351257, 11471230, 11501153, 11561131, 12314335, 12321299, 12411269, 12411493, 12411829, 12411930, 12412055, 12511029, 12511031, 12511033, 12511072, 12511145, 44441035
Subjects with AEs missing in the CRF file, yet present in ADAE: 10071441, 10081152, 10131229, 10131386, 10131718, 10161087, 10191021, 10191145, 10211081, 10211084, 10281083, 10281205, 10381050, 10441163, 10471012, 10551128, 10551139, 10551145, 10551182, 10791183, 10801013, 10811179, 10831050, 10831173, 10841219, 10841538, 10871266, 10901140, 10911213, 10911247, 10911297, 10931067, 10931128, 11071191, 11091036, 11091074, 11091164, 11091204, 11101236, 11311140, 11331537, 11401066, 11401244, 11401282, 11401285, 11411143, 11461181, 11461302, 11521053, 11521095, 11521316, 11561001, 11781257, 11951017, 11951023, 12261477, 12312660, 12315186, 12315579, 12411053, 12411117, 12411347, 12411410, 12411561, 12411568, 12412191, 12412218, 12511060, 12541109, 12541189, 12601018, 12601108, 44441422
ECFR.gov - 21 CFR 312.57 - https://www.ecfr.gov/current/title-21/chapter-I/subchapter-D/part-312/subpart-D/section-312.57
Ema.europa.eu - ICH guideline for good clinical practice E6(R2) - Step 5 - https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-good-clinical-practice-e6r2-step-5_en.pdf
Documentation - adae_to_covid_symptoms.csv - https://github.com/OpenVaet/pfizer_docs_R/blob/main/adae_to_covid_symptoms.csv
PHMPT.org - 125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, Page 97
R Script - aes_and_symptoms_by_subjects.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/aes_and_symptoms_by_subjects.R
R Script - unblinding_by_arm_and_age_groups.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/unblinding_by_arm_and_age_groups.R
International Journal of Vaccine Theory, Practice, and Research - Corinne Michels et al. - Forensic analysis of the 38 subject deaths in the 6-Month Interim Report of the Pfizer/BioNTech BNT162b2 mRNA Vaccine Clinical Trial - https://ijvtpr.com/index.php/IJVTPR/article/view/86
The New England Journal of Medicine - Fernando P. Polack et al. - Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine - https://www.nejm.org/doi/full/10.1056/nejmoa2034577
Centers for Disease Control & Prevention - Underlying Cause of Death, 2018-2022, Single Race Request - https://wonder.cdc.gov/ucd-icd10-expanded.html, "Year/Month: 2020; 2021", "Group By: State; Five-Year Age Groups; Year; Month", "Show Totals: Disabled", "Show Zero Values: False", "Show Suppressed: False", "Calculate Rates Per: 100,000", "Rate Options: Default intercensal populations for years 2001-2009 (except Infant Age Groups)"
Documentation - Underlying Cause of Death, 2018-2022, Single Race.txt - https://raw.githubusercontent.com/OpenVaet/pfizer_docs_R/main/us_mortality/Underlying%20Cause%20of%20Death%2C%202018-2022%2C%20Single%20Race.txt
Documentation - Single-Race Population Estimates 2020-2022.txt - https://github.com/OpenVaet/pfizer_docs_R/blob/main/us_mortality/Single-Race%20Population%20Estimates%202020-2022.txt
Centers for Disease Control & Prevention - Single-Race Population Estimates 2020-2022 Request - https://wonder.cdc.gov/single-race-v2022.html, "Yearly July 1st Estimates: 2020; 2021", "Group By: State; Yearly July 1st Estimates; Age Group", "Show Totals: True", "Show Zero Values: False"
R Script - model_deaths_to_us_underlying_mortality.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/model_deaths_to_us_underlying_mortality.R
NEJM.org - Protocol - https://www.nejm.org/doi/suppl/10.1056/NEJMoa2034577/suppl_file/nejmoa2034577_protocol.pdf, Page 174
European Medicine Agency - Committee for Medicinal Products for Human Use (CHMP) - Product Assessment report (PAR) - https://www.ema.europa.eu/en/documents/assessment-report/comirnaty-epar-public-assessment-report_en.pdf, Page 18
Pharmaceutical and Medical Products Agency - Pharmaceutical Evaluation Division, Pharmaceutical Safety and Environmental Health Bureau, Ministry of Health, Labour and Welfare - Report on the Deliberation Results - https://www.pmda.go.jp/files/000243206.pdf, Page 13
PHMPT.org - Protocol C4591001 Protocol Amendment 9, 29 October 2020 - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 310
PHMPT.org - Protocol C4591001 Protocol Amendment 9, 29 October 2020 - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 360
PHMPT.org - Protocol C4591001 Protocol Amendment 9, 29 October 2020 - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 360
Australian TGA, FOI 2389 - Pfizer/BioNTech COVID-19 mRNA vaccine (BNT162, PF-07302048) TGA Pre-Submission Meeting - https://www.tga.gov.au/sites/default/files/foi-2389-03-1.pdf, Pages 21 to 23
Australian TGA, FOI 3659 - BNT162b2 (PF-07302048) Comparability Report for PPQ Drug Product Lots - https://www.tga.gov.au/sites/default/files/2022-08/foi-3659-04.pdf, Pages 4 & 5
Australian TGA, FOI 3659 - BNT162b2 (PF-07302048) Comparability Report for PPQ Drug Product Lots - https://www.tga.gov.au/sites/default/files/2022-08/foi-3659-04.pdf, Page 3
PHMPT.org - 125742_S2_M3_32p5_batch-analyses.pdf, BNT162b2 3.2.P.5.4. Batch Analyses - https://phmpt.org/wp-content/uploads/2023/11/125742_S2_M3_32p5_batch-analyses.pdf, Page 3
PHMPT.org - 125742_S1_M5_5351_c4591001-interim-mth6-patient-batches.pdf - https://phmpt.org/wp-content/uploads/2022/06/125742_S1_M5_5351_c4591001-interim-mth6-patient-batches.pdf
Documentation - The Vaccine Adverse Event Reporting System (VAERS).txt - https://github.com/OpenVaet/pfizer_docs_R/blob/main/vaers/The%20Vaccine%20Adverse%20Event%20Reporting%20System%20(VAERS).txt
Centers for Disease Control & Prevention - VAERS - https://wonder.cdc.gov/vaers.html, VAERS Data Search, “Group by:Year Reported,Month Reported, Vaers Id”, “Vaccine Products: COVID19 (COVID19 VACCINE), COVID19-2 (COVID19-2)”, “Vaccine Manufacturer: Pfizer\BioNTech”, “Vaccine lot:EE8493, EJ0553”
Documentation - The Vaccine Adverse Event Reporting System (VAERS)_other_batches.txt - https://github.com/OpenVaet/pfizer_docs_R/blob/main/vaers/The%20Vaccine%20Adverse%20Event%20Reporting%20System%20(VAERS)_other_batches.txt
PHMPT.org - 125742_S1_M5_5351_c4591001-fa-interim-patient-batches.pdf - https://phmpt.org/wp-content/uploads/2022/06/125742_S1_M5_5351_c4591001-fa-interim-patient-batches.pdf
R Script - process_2_subjects_by_sites_and_date.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/process_2_subjects_by_sites_and_date.R
Documentation - process_2_recipients_by_dates_and_sites.csv - https://github.com/OpenVaet/pfizer_docs_R/blob/main/process_2_recipients_by_dates_and_sites.csv
PHMPT.org - 125742_S1_M5_5351_c4591001-interim-mth6-randomization-sensitive.pdf, 16.1.7.1 Listing of Randomization Scheme and Actual Vaccine Received – All Subjects ≥16 Years of Age - https://phmpt.org/wp-content/uploads/2022/05/125742_S1_M5_5351_c4591001-interim-mth6-randomization-sensitive.pdf
R Script - process_2_subjects_by_randomization_numbers.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/process_2_subjects_by_randomization_numbers.R
R Script - compare_process_2_identification_methods.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/compare_process_2_identification_methods.R
R Script - model_process_2_aes.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/model_process_2_aes.R
MHRA’s Public Assessment Report for the Authorisation for Temporary Supply of Pfizer/BioNTech’s COVID-19 mRNA Vaccine BNT162b2 - https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1112667/COVID-19_mRNA_Vaccine_BNT162b2__UKPAR___PFIZER_BIONTECH_ext_of_indication_11.6.2021.pdf
GOV.UK - ARCHIVE: Information for Healthcare Professionals on COVID-19 Vaccine Pfizer/BioNTech (Regulation 174) - https://www.gov.uk/government/publications/regulatory-approval-of-pfizer-biontech-vaccine-for-covid-19/information-for-healthcare-professionals-on-pfizerbiontech-covid-19-vaccine
Note that the PAR from December 2020 states a lymphadenopathy rate from doses 1 and 2 of 0.3% but the updated information for healthcare professionals cites a rate of 0.4%. The difference is because the original clinical trial had not finished recruiting participants when authorization was granted in December, 2020. The complete trial data put the rate at 0.4% instead of 0.3%.
European Journal of Nuclear Medicine and Molecular Imaging - Cohen, D., Hazut Krauthammer, S., Wolf, I. et al. - A sigh of relief: vaccine-associated hypermetabolic lymphadenopathy following the third COVID-19 vaccine dose is short in duration and uncommonly interferes with the interpretation of [18F]FDG PET-CT studies performed in oncologic patients. -
https://doi.org/10.1007/s00259-021-05579-7
The New England Journal of Medicine - Moreira ED Jr, Kitchin N, Xu X, et al. - Safety and efficacy of a third dose of BNT162b2 Covid-19 vaccine. -
https://www.nejm.org/doi/full/10.1056/NEJMoa2200674, Table 2
PHMPT.org - 125742_S1_M5_CRF_c4591001-1223-12231058.pdf - https://phmpt.org/wp-content/uploads/2023/05/125742_S1_M5_CRF_c4591001-1223-12231058.pdf
R Script - aes_cutoff_anomalies.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/aes_cutoff_anomalies.R
PHMPT.org - 125742_S1_M5_CRF_c4591001-1226-12261769.pdf - https://phmpt.org/wp-content/uploads/2023/06/125742_S1_M5_CRF_c4591001-1226-12261769.pdf
PHMPT.org - 125742_S1_M5_CRF_c4591001-1231-12312420.pdf - https://phmpt.org/wp-content/uploads/2023/06/125742_S1_M5_CRF_c4591001-1231-12312420.pdf
PHMPT.org - 125742_S1_M5_CRF_c4591001-1231-12313610.pdf - https://phmpt.org/wp-content/uploads/2023/06/125742_S1_M5_CRF_c4591001-1231-12313610.pdf
PHMPT.org - 125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, Page 206
PHMPT.org - 125742_S1_M5_CRF_c4591001-1079-10791044.pdf, https://phmpt.org/wp-content/uploads/2023/08/125742_S1_M5_CRF_c4591001-1079-10791044.pdf, Page 160
PHMPT.org - 125742_S1_M5_CRF_c4591001-1117-11171146.pdf, https://phmpt.org/wp-content/uploads/2023/05/125742_S1_M5_CRF_c4591001-1117-11171146.pdf, Page 178
R Script - inspect_menstrual_disorders_aes.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/inspect_menstrual_disorders_aes.R
MDPI, Medicina - Nahid Ibrahim Fallatah et al. - Menstrual Changes Following COVID-19 Vaccination: A Cross-Sectional Study - https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10890281/
Journal of Infection and Public Health - Hanan M. Al Kadri et al. - COVID-19 vaccination and menstrual disorders among women: Findings from a meta-analysis study - https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9979695
R Script - parse_medical_history.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/parse_medical_history.R
R Script - positive_pcrs_by_days_from_start.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/positive_pcrs_by_days_from_start.R
R Script - tests_on_symptoms.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/tests_on_symptoms.R
R Script - tests_on_symptoms_arg.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/tests_on_symptoms_arg.R
R Script - tests_on_symptomatic_visits.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/tests_on_symptomatic_visits.R
PHMPT.org - 125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, Page 63
PHMPT.org - 125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf - https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, Page 97
R Script - tests_on_symptomatic_visits_4_days.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/tests_on_symptomatic_visits_4_days.R
R Script - tests_on_symptomatic_visits_by_sites.R - https://github.com/OpenVaet/pfizer_docs_R/blob/main/tests_on_symptomatic_visits_by_sites.R
Thank you to all the 8 authors who contributed in putting this together. Addressing these anomalies will be a challenge, and I'm hopeful this prompts a thorough audit of both the sites and the CROs.
All of the above is true, however, the problem was not lack of regulatory oversight. These products were taken out of the pathway of regulatory oversight for which drugs and biologics are subject. They were purposely contracted by the DoD under OTA, and were legally defined not as drugs or vaccines but as "medical countermeasures." This is important to understand. There are laws on the books that enable the DoD to contract for medical countermeasures to be used in a declared medical emergency. As such a "medical countermeasure" is in the category of weapon, or any other product the DoD might employ in the defense of the country or in war. There was never any intention to treat these substances as if they were actually vaccines or drugs. The FDA cannot regulate a weapon that the DoD has purchased. If you are serious about legal challenges you need to understand the laws that have enabled the US bioterrorism program. Katherine Watt has undertaken a very detailed study of all these laws, and why "regulatory oversight" grounds will never succeed in a court of law. In fact it is part of a coverup to focus on regulation. You are participating in covering up the fact that the DoD and Congress created a bioterrorism program that can be used on Americans and others around the world. Watt's substack is: www.bailiwicknews.substack.com.