


Comment déterminer la vérité sur les origines du Covid-19, une visite guidée de BLAST
Utiliser BLAST est facile. Je vais vous montrer à quel point, et comment prouver que le SARS-Cov-2 est fabriqué par l’homme.

L’article ci-dessous est une traduction française de l’article d’Arkmedic “How to BLAST your way to the truth about the origins of COVID-19”. Il constitue une vue d’ensemble claire des raisons pour lesquelles il devrait être transparent que le Covid-19 a été fabriqué par l’Homme, aussi l’avons-nous traduit à destination du public français.
J’ai eu l’intention d’écrire cet article depuis une éternité. Du moins, depuis que Prashant Pradhan (un merveilleux, honnête et courageux scientifique en génomique) a soulevé en Février 2020 l’hypothèse que le virus SARS-Cov-2 ait été fabriqué en laboratoire. Nous avons vu depuis de nombreuses preuves que le virus avait été fabriqué en labo, l’une des meilleures étant ici sur zenodo, dotée de sa propre vidéo pour les non-familiers des courbes bayésiennes. A l’heure où j’écris cet article, ces liens sont toujours en ligne depuis 12 mois, ce qui est plutôt remarquable pour tout article qui ose défier le narratif de propagande de notre bien aimée « presse libre [sponsorisée par l’industrie pharmaceutique] ».
Enfin…
BLAST est le répertoire du NCBI/NIH (autrement dit du gouvernement américain) pour les séquences génomiques et protéomiques, entre autres choses. C’est là que tous les génomes sont déposés, par les scientifiques du monde entier, lorsqu’ils font une découverte. Sa principale fonction est de permettre la recherche et la comparaison d’une séquence génétique que vous pourriez rencontrer lors d’une expérience. Qu’est-ce qu’une séquence génétique ? C’est facile. C’est une ligne de code, faite de n’importe quelle combinaison de 4 lettres dans une séquence. Vous vous souvenez du film GATTACA ? Si vous ne l’avez pas encore vu, vous devriez – c’est une autre dystopie qui commence à ressembler un peu trop à la réalité.
 - AZ Movies")
Le titre du film est basé sur les 4 bases de nucléotides (G, A, T, C) qui composent le code génétique de chaque ADN humain. Il y a environ 3 milliards de celles-ci dans chaque cellule, composant un code unique, dont vous êtes – individu unique – le résultat ! Le code fonctionne par paires, donc G-C et A-T sont toujours combinés pour former une double-hélice que vous pouvez voir dans l’affiche du film, de sorte que GATTACA sur un brin serait TGTAATC sur l’autre (le complément inversé). Une des bonnes choses avec BLAST et qu’il n’est pas conditionné par la version que vous lui fournissez, et vous redirigera vers le bon gène.
Un autre point à noter à ce stade ; les probabilités. Vous les avez pratiquées à l’école avec par exemple des lancers de pièce (où le code est F pour face et P pour pile). La probabilité de FFFF serait 1 sur 2 ^ 4 = 1/16. La même chose s’applique pour PPPP. La même chose s’applique pour PPTP, ou n’importe quelle séquence spécifique de 4 lancers. Essayez vous-mêmes si vous avez des doutes (prédisez une séquence et voyez combien de fois vous avez besoin de lancer votre pièce en moyenne). Le code génétique est essentiellement une « pièce à quatre cotés ». Donc pour chaque séquence génétique (par exemple GATC) la probabilité d’obtenir cette séquence exacte est d’une sur 4 ^ 4, où pour chaque nombre N de nucléotide (nt ou bases) la chance est d’une sur 4 ^ N (une simplification, puisque dans des situations données la probabilité de la prochaine base d’être X dépend des bases qui l’entourent).
BLAST a deux catégories – nucléotides (BLASTn) et protéines (BLASTp). BLASTp s’occupe des séquences d’acides aminées, exactement de la même façon qu’il procède pour les séquences de nucléotides. Mais il y a une grosse différence : il y’a plus de 20 amino-acides (plutôt que 4 nucléotides) et par conséquent chaque séquence courte (par exemple QTNS = Glu-Hr-Asn-Ser) aurait une probabilité (simplifiée) aux alentours de 1 sur 20 ^ 4 ce qui est 1 sur 160 000. La probabilité d’une séquence spécifique de 5 amino-acides survenant par chance, sur la même base, s’envole à une sur 3.2 millions !

Donc, pressez le bouton “Protein BLAST”, et voilà l’écran suivant que je vais vous décrire.

Dans la partie « 1 » vous devez entrer la séquence d’amino-acide qui vous intéresse (BLAST ajoute « >unnamed potein product » automatiquement). Par chance vous n’avez pas besoin d’approfondir trop avant cette partie, puisque nous nous concentrerons seulement sur 4 séquences contenues dans le génome/protéome du virus SARS-Cov-2, et celles-ci ont été exposées dans le merveilleux papier de Prashant Pradhan « Similitudes étranges des inserts uniques dans la protéine de pointe 2019-nCoV avec la gp120 et le gag du VIH-1 », publié le 31 janvier 2020, quelques jours après que la séquence virale ait été publiée.
La section dont vous avez besoin est la table 1 que je republie ici, et sur laquelle vous pouvez constater que j’ai surligné la séquence de 6 amino-acides TNGTKR dans la cellule [1] marquée en rouge sur l’écran de BLASTp.

Pour la partie [2] de l’écran de saisie de BLASTp, vous aurez besoin de définir quelques filtres. Le premier est de restreindre la recherche aux « viridaes (virus) » (ou vous pouvez juste entrer 10239 qui est l’identifiant de taxinomie). La raison pour laquelle nous opérons cette restriction est qu’il y’a des milliards d’espèces sur la planète et que BLAST va effectuer sur sa recherche sur toute sa base de connaissance ; or ce qui nous intéresse ici est uniquement de savoir de quel virus ce motif provient. Vous n’êtes pas particulièrement intéressé de savoir si le motif se trouve chez un poulpe ; bien qu’il soit possible qu’un poulpe soit rentré en interaction avec la fameuse chimère de pangolin et de chauve-souris dont nous parlent Peter Daszak, Dominic Dwyer & leurs disciples, ce qui en ferait un ménage à trois zoonotiques, nous essaierons ici de rester dans le domaine du réel.
Le second filtrage est d’exclure toutes les références à SARS-Cov-2 qui ont déjà été accumulées dans la base de données, puisque toutes celles-ci vont apparaitre (des milliers d’entre eux) et nous ne sommes pas intéressés.
Une fois que vous avez entré ces filtres, pressez le bouton « BLAST » et qu’obtenez-vous ? Vous obtenez une liste de candidats qui ont une homologie proche de cette séquence. Parce que c’est une séquence très courte l’homologie devrait être de 100%. Le haut de la page sera un résumé de ce que vous avez requis, et le reste de la page sera composé des séquences correspondantes. Ce que vous allez voir immédiatement est que le sommet de la liste correspond à deux virus synthétiques qui sont des chimères de SARS-Cov-2, et un autre virus, qui est sorti il y’a deux ans d’un laboratoire (parce que nous n’en avons pas assez). Les entrées suivantes dans la liste correspondent à une vaste quantité de références au VIH-1 (ndt : HIV-1 en anglais).

Vous pouvez cliquer sur n’importe laquelle de ces entrées ; vous serez amené à un écran d’alignement, ou l’alignement entre le sujet (votre TNGTKR) et la requête (tous les virus) est montré, et où vous verrez si vous naviguez en bas de la page que l’alignement n’est confirmé que pour VIH-1, jusqu’à ce que vous voyiez d’autre protéines synthétiques ou hypothétiques, puis enfin un autre virus, VIH-2…
D’accord, mais une correspondance pourrait être une coïncidence. Dans cette liste vous verrez une probabilité estimée « E-value » qui est un indicateur de la probabilité de trouver une correspondance telle que la séquence requise, et cette probabilité devrait être aussi proche de zéro que possible. Ici, la probabilité est à 282, ce qui ne reflète guère que la probabilité d’obtenir une correspondance pour une séquence très courte.
Donc voilà le hic. Soit ces séquences vont, par chance, correspondre à toute une série d’autres virus (parce que la probabilité estimée est si haute, et que par conséquent nous devrions avoir beaucoup de correspondances) ou bien ce seront des séquences vraiment inhabituelles qui sont spécifiques, préférentielles ou uniques au VIH-1. Comment devrions-nous adresser cette question ? Et bien passons à la prochaine séquence, HKNNKS, qui est une autre séquence courte. Pour écrémer un peu les résultats nous pouvons sélectionner un filtre pour assurer que 100% de la séquence corresponde (en haut à droite de l’écran). Maintenant souvenez-vous que si la correspondance précédente avec le VIH-1 était aléatoire, nous ne devrions pas voir de nouvelle correspondance sur cette liste, parce qu’elle serait précédée par toutes les centaines de virus qui correspondraient davantage. Oups…

Notez les autres correspondances – le RaTG13 de la chauve-souris qui n’est apparu dans la base de données qu’après que les gens aient commencé à poser des questions sur l’origine du Coronavirus, et qui est probablement une séquence synthétique, et d’autres virus synthétiques apparus après l’émergence du SARS-Cov-2. Donc, le VIH-1 est le SEUL virus antérieur qui corresponde à ces deux séquences.
Passons à la 3e séquence. C’est une séquence plus longue. RSYLTPGDSSSG. Je me demande quel virus (ou série de virus aléatoires) ceci va correspondre… Oh, regardez…

Donc, dans cette recherche, nous avons restreint la correspondance à 100% pour éliminer le bruit et des hypothétiques protéines. Il ne nous reste que les virus synthétiques post-covid, et RaTG13 (lui aussi post-covid). Le seul virus restant dans la liste est, vous l’aurez deviné, VIH-1. Quelles sont les chances que VIH-1 apparaissent dans les trois recherches ?
Et pour compléter cette démonstration en 4 temps nous aurons besoin de la 4e séquence identifiée dans l’article de Pradhan, qui est QTNS——PRRA. C’est une séquence très intéressante parce que – nous y reviendrons – c’est la séquence du site de clivage de furine. C’est intéressant parce que les coronavirus de type béta comme le SARS-Cov-2 n’ont pas de site de clivage de furine, il est le seul à avoir cette particularité. A coup sûr ce site ne pourrait avoir son origine dans VIH-1 ? Et bien, il ne vient pas de la protéine GP120 comme les trois autres séquences, et est complètement différent et à un autre emplacement du virus, comme nous allons le montrer rapidement, mais pour l’instant lançons BLASTp.
Cette fois les résultats sont un peu plus brouillons parce que nous avons une quantité de protéines hypothétiques et synthétiques qui ont été ajoutées depuis que Sars-Cov-2 a été libéré (j’aurais dû écrire cet article l’an dernier). Mais VIH-1 apparait à nouveau dans la liste et cette fois je vais juste montrer les alignements entre la protéine gag et le coronavirus – dans ce cas il y’a une suppression depuis la protéine du VIH-1.
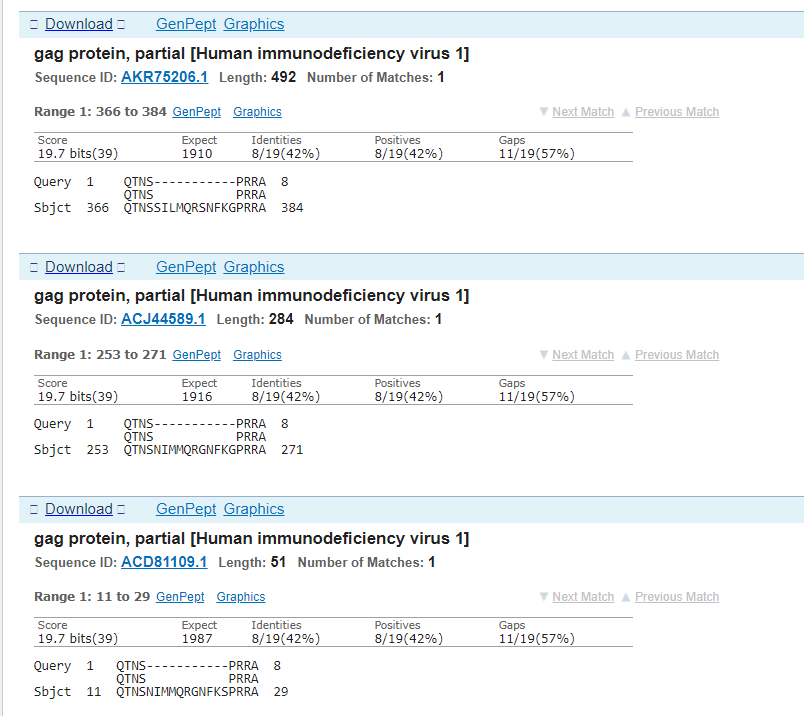
Donc, nous avons 4 correspondances avec des séquences du VIH, et qui ne correspondent à aucun autre virus (à l’exclusion des synthétiques créés à postériori). Quelles sont les chances ? Proches de zéro.
Mais prêtons attention à un autre détail. Il ne s’agit pas de séquences aléatoires du VIH. Dans son article, Pradhan a été au-delà et a recréé la structure du virus avec la position de ces quatre insertions. Admirez, ces insertions « aléatoires » - toutes issues du VIH – sont toutes aux sites de liaison du Coronavirus. Quelles sont les chances ?

A ce stade, il est possible que vous ne soyez pas convaincu. Malgré le fait que le VIH-1 soit le seul virus qui apparaisse dans la liste avec nos quatre insertions, parmi les centaines de milliers de virus en circulation. Et que le VIH-1 devrait n’avoir aucune chance de former des virus recombinants avec des coronavirus de chauve-souris dans la nature, et aucune chance réelle de former 4 recombinaisons différentes qui se retrouvent par hasard aux sites de fusion du virus. Mais si ça ne vous a pas convaincu, voici une particularité de ces quatre insertions à laquelle nous devons prêter attention.
Nous allons revenir aux nucléotides, les G-A-C-T qui forment les séquences qui codent les amino-acides dont nous avons parlé plus avant. La référence d’origine du Coronavirus dans Genbank est NC_045512.1 et peut être trouvée ici : https://www.ncbi.nlm.nih.gov/nuccore/NC_045512.1
Vous pouvez vous amuser avec le génome de la séquence grâce à BLASTn (pour les nucléotides), en vous rendant sur sa page et en sélectionnant « Run BLAST » depuis la colonne de droite, qui vous emmènera à une page BLAST similaire à celle utilisée précédemment pour les protéines.
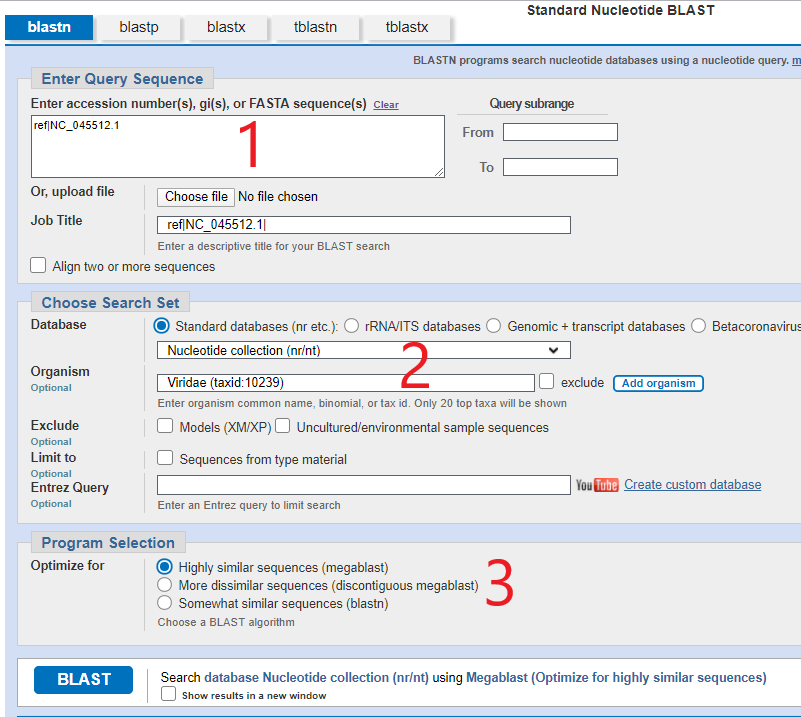
D’une façon similaire, vous allez vouloir saisir NC_045512.1 (ou la mise à jour NC_045512.2) dans la première fenêtre, choisir les options montrées en partie marquée [2], sélectionner « Mégablast » en partie 3, et cliquer « Blast ». Vous obtiendrez une liste de génomes de Sars-Cov-2 qui correspondent (évidemment). Je ne vais pas vous montrer cet écran parce qu’il n’a aucune importance en l’espèce, mais voici l’écran que vous obtenez si vous regardez de plus prêt deux séquences proches et que vous cliquez « CDS feature » pour superposer les séquences amino-acides. Vous vous retrouverez avec des pages de résultats qui ressemblent aux suivants :

Dans cette section spécifique vous pouvez voir qu’il s’agit d’une séquence de « protéine spike » (glycoprotéine de surface) et les nucléotides sont labélisées 23538 … 23771 (environ 30 000 nucléotides ou bases, c’est-à-dire G-C-A-T). [N.B : il s’agit en fait de RNA donc vous devriez avoir un U à la place de chaque T, mais BLAST compense automatiquement ce facteur par soucis de simplicité]. Le nombre plus faible est le nombre d’amino-acides dans la séquence de la protéine de sorte que pour chaque trois nucléotides, le nombre augmente de 1 amino-acide. Le surlignage est l’amino-acide 677 (Q) à 686 (S), résultant en 677-> 686 = QTNSPRRARS.
Maintenant, ceci est vraiment intéressant, parce que non seulement nous avons vu que cette section QTNS est dérivée du VIH, mais il y’a aussi quelque chose de très spécial à propos de la séquence adjacente PRRAR, parce qu’il s’agit d’un site de clivage de furine, et que comme nous l’avons vu, celles-ci n’existent pas dans ce type de virus de type SARS. C’est une insertion de séquence génomique virale, mais personne ne sait vraiment comment elle est arrivée là (un mystère similaire aux séquences de VIH). Afin de voir d’où elle provient nous avons besoin de regarder au-delà de la séquence d’amino-acides et de retourner à la séquence du génome.
La séquence du génome que l’on peut observer pour cette séquence amino-acide est :
CAGACTAATTCTCCTCGGGCGGGCACGTAGT, qui est un code de 30 nucléotides pour 10 amino-acides. Les chances que cette séquence survienne par chance serait de l’ordre de l’infinitésimal – elle a donc dû apparaitre quelque part (c’est-à-dire dans un autre virus) ou alors une partie doit-être synthétique. Donc « BLASTons » la, et cette fois excluons les « constructions synthétiques » (Synthetic Constructs » de notre recherche - parce que nous cherchons de vrais virus, pas des synthétiques). Qu’obtenons-nous ?

A ce stade vous avez l’habitude de ces rapports … nous pouvons constater que les seules séquences virales ici présentes sont synthétiques, et si vous cliquiez sur chacune d’entre elles vous pourriez voir que leur enregistrement date de Février 2020. En d’autres mots, aucun virus existant n’a cette séquence génétique. Ce qui est étrange, parce que pour qu’un virus acquiert une aussi longue séquence telle que celle-ci il doit l’hériter d’un autre organisme. Il n’a pas de laboratoire à disposition pour manipuler sa séquence génétique, et les chauves-souris non plus (d’où la petite plaisanterie de Jikky, la souris de laboratoire) …
Il est relativement facile de changer un seul nucléotide (un « simple point de mutation » ou « SNP ») ou même d’insérer ou supprimer des nucléotides (ce qui est moins commun) mais d’insérer 20 ou 30 nucléotides et d’obtenir un code qui fonctionne ? Non, une telle mutation doit provenir d’un autre virus ou a été faite en laboratoire.
Donc, d’où provient ce code ? Et bien il s’avère que BLAST peut nous dire, avec un certain degré de certitude, d’où provient une partie de ce code, particulièrement la partie qui code la section PRRAR (le site de clivage de la furine qui est unique).
La partie qui nous intéresse est dans l’indice fourni par Jikky.
CTCCTCGGCGGGCACGTAG. « BLASTons » -la.

Ce que vous voyez est la même séquence (mal classifiée SARS-Cov-2) pour les 9 premiers résultats, et ensuite aucun des résultats n’a une correspondance de 19/19. Ce que ça signifie est qu’il n’y a aucun virus connu de l’homme qui ait cette séquence de génome particulière, avant la découverte de SARS-Cov-2. Donc d’où diable a-t-elle pu provenir ? Pour répondre à cette question vous aurez besoin d’une autre base de données. Retournons à l’écran de requête de BLASTn et changeons l’option de base de données ciblée vers « séquences brevetées » (Patent sequences (pat)). Retirez toutes les exclusions et lancez BLAST.

Ces résultats doivent être un peu expliqués car le haut de la liste comprend les résultats des brevets de cette année. Ils sont préfixés XO2021 et WO2020 et peuvent donc être ignorés. Juste en dessous, nous trouvons les résultats qui nous intéressent. J’ai seulement souligné les trois premiers mais la liste complète de ces nombreux brevets appartient à la même société, et il vous suffit de cliquer sur le numéro d’accession à droite pour accéder aux détails.

Accédons donc à ces détails, et voyons quelle société, que nous connaissons tous (maintenant), est une entreprise pharmaceutique qui n’a jamais produit un médicament autorisé mais qui a une capitalisation boursière de plus de 80 milliards de dollars…

En effet. Chacun de ces brevets qui contient la séquence de 19 nucléotides (dont la chance d’apparition par chance est inférieure à 1 sur 1 milliard) appartient à Moderna. [Notez que la séquence est en fait la séquence complémentaire inverse, mais qui est le résultat direct de la lignée cellulaire dans laquelle elle s’est produite : la lignée cellulaire mutée MSH3 conçue pour développer des vaccins contre le cancer, le brevet Moderna concernait en fait un gène muté à cette fin MSH3.]
Pour que cette séquence soit apparue dans ce virus, le virus qui a été fabriqué avec des inserts de VIH, devait avoir été infecté dans des lignées cellulaires brevetées fournies par Moderna qui avaient cette séquence unique qui n'a été vue dans aucun autre virus.
En théorie, rien n’est impossible en Science, médecine ou génomique. Un virus SARS qui émerge naturellement, avec 3 insertions de VIH à ses sites de liaison, et qui contient aussi un site de clivage de furine qui n’existe pas dans la nature, mais existe dans un brevet Moderna… Penser que ça puisse arriver par chance est une hypothèse sérieusement folle. Un éléphant rose volant serait des millions de fois plus probable.
[Mise à jour du 21 Février 2022… L’article que l’on attendait est maintenant publié, et a confirmé que la séquence de 19 nucléotides provient d’un brevet Moderna, comme l’on en discutait plus haut. Il vaut la peine d’être lu si vous avez le temps.]
[Mise à jour du 4 Décembre 2022… J’aurais dû ajouter précédemment des liens versle long article de Charles Rixey sur les documents sous-jacents à ce "Mythe de l'horloger aveugle" ainsi que l'article de suivi (partie 2) "La preuve absolue : Les séquences GP120" qui sont maintenant ajoutés ci-dessous, en plus de l'excellent résumé d'Igor Chudov "Sars-Cov-2 was lab made under project DEFUSE"]


Hoping this makes the French establishment sit up and take notice of you and Arkmedic